- 首页 > 扩增子/宏基因组测序
扩增子/宏基因组测序
新冠病毒感染之后的生理生化指标,一直是研究人员非常关注的方向。而同样作为微生物,环境、人体肠道等来源的微生物也会对宿主机体产生或好或坏的影响。在病毒感染的背景下,肠道菌群的变化、环境微生物的差异都会为我们的诊断和治疗带来帮助。
扩增子和宏基因组是微生物高通量测序的常用技术手段,能快速、便捷且高通量地研究复杂环境样本中的微生物组成,辅助寻找新冠病毒感染后的标志性微生物组成变化,甚至利用宏基因组技术,探寻关键功能基因,帮助研究人员深入了解新冠病毒所带来的危害,实现精准防控。
研究方案一
1. 研究背景
病毒侵染宿主后,对宿主的肠道菌群、呼吸道菌群等微生物环境造成影响,进而产生不同的生理生化反应。通过不同患病阶段样本、不同患病程度样本等的比较分析,寻找微生物菌群的多样性变化,找到数据特征,阐述生物学问题。
2. 技术策略
1)扩增子测序研究不同患病阶段或患病程度的肠道/呼吸道/口腔等菌群多样性。
2)宏基因组测序研究不同患病阶段或患病程度的肠道/呼吸道/口腔等菌群多样性,并深入挖掘重要菌种和功能基因。
3. 实验设计
|
分组 |
患者数 |
采样点 |
样本类型 |
样本量 |
|
重症组 |
≥30 |
疾病中/治愈后 |
粪便样本/咽拭子/口腔唾液等其他微生物样本 |
各1例/患者/时间点 |
|
轻症组 |
≥30 |
疾病中/治愈后 |
||
|
无症状患者组 |
≥30 |
隔离群体/解除隔离后 |
||
|
正常组 |
≥30 |
未接触病例 |
注:其他医学分类或不同患病时间点也可以。
4. 测序策略
扩增子测序:
推荐的测序平台:Illumina HiSeq2500平台
推荐的扩增区域:单V4区、双V3V4区等,如有其它需求请联系华大当地销售
推荐的测序数据量:单个样本测序数据量不低于50,000 tags
宏基因组测序:
推荐的测序平台:MGISEQ平台
推荐的测序读长: PE150
推荐的测序数据量:单个样本测序数据量不低于5 Gb
5. 案例展示
5.1 案例解析一
中文名:肠道菌群代谢与HIV感染炎症之间的相互作用
英文名:Interplay between gut microbiota metabolism and inflammation in HIV infection
发表期刊:The ISME Journal
影响因子:9.520
发表时间:2018年4月
研究背景:HIV感染导致肠道相关淋巴组织的破坏,使得肠道菌群的组成发生变化。但目前还缺乏关于HIV感染如何影响微生物与宿主之间相互作用的研究。本文结合宏基因组和宏转录组数据来研究HIV相关的微生物群落变化。结果表明,HIV相关微生物一方面很好地适应炎症环境,例如抗氧化应激反应通路的高表达、抗炎反应过程的低表达,另一方面促进肠道炎症的发生发展。本文通过共发生网络和代谢网络,分析了群落结构中物种标记物和代谢标记物的相关性。通过贝叶斯网络,发现了维持群落结构稳定性的关键通路。此外,确定了群落中各物种对代谢活动的贡献,以及各物种和宿主健康的相互作用。
研究对象:病毒血症患者12例(VU=12),免疫应答者18例(IR =18),免疫无应答者9例(INR=9),以及未感染HIV的健康对照15例(HIV-=15)。
研究方法:宏基因组、宏转录组、代谢组学
主要结果:
1) HIV+组和HIV-组样本中KO基因含量的差异性很大。HIV+样本菌群表现为促炎症反应通路(ko00540、ko05111、ko05120)的增加,抗氧化应激反应通路(ko00250、ko00908)的增加,而信号转导和膜转运通路的减少。HIV+样本菌群表现为Prevotella、Acidaminococcus、Streptococcus等菌属的增加,而Bacteroides、Bifidobacterium、Akkermansia、Odoribacter、Alistipes等菌属的减少。
2)宏转录组数据结果表明,HIV+组和HIV-组样本中RNA-KO基因含量的差异性很大。HIV+样本菌群表现为应激反应通路(ko04141、ko00521、ko00730、ko00053)的转录本丰度增加,而抗炎症代谢过程(ko00650、ko00640、ko00071)的丰度减少。基于宏转录组数据的物种注释结果,HIV+样本菌群表现为Prevotella、Acidaminococcus、Coprobacillus、Streptococcus等菌属的增加。这些被认为是转录活跃细菌。
3)使用广义线性模型GLM分析,发现HIV+组中细菌物种标记物与通路标记物为正相关。发现物种标志物提供了涉及其相关代谢通路的基因,而且普氏菌属各个菌种都提供了涉及HIV相关通路的基因,表明普氏菌属在HIV发病过程中的重要作用。
4)物种关系网络图产生了20个模块,各模块都包含3个以上物种,最大模块包含34个物种。大多数模块的主导细菌为厚壁菌门与拟杆菌门,少数模块的主导细菌为放线菌门和变形菌门。结果还发现,如果模块中包含双歧杆菌属(放线菌门),则该模块中不存在变形菌门,推测放线菌门和变形菌门有竞争关系。
5.2 案例解析二
中文名:16S扩增子测序研究H1N1感染后的呼吸道菌落
英文名:16S rDNA sequencing analysis of upper respiratory tract flora in patients with influenza H1N1 virus infection
发表期刊:Frontiers in Laboratory Medicine
影响因子:1.02(2018)
发表日期:2017
研究背景:呼吸黏膜上的菌落组成跟人体疾病的产生和发展有着千丝万缕的关系。健康的菌落组成可以参与调节流感病毒侵染时的免疫反应,提高免疫耐受力。人在感染H1N1流感病毒后,病程进展很快,且容易发生继发性的严重的细菌感染,进而促使H1N1病毒在下呼吸道发生复制,产生严重的后果。16S扩增子测序可以快速了解样品中微生物的分类和系统发育,因此,作者通过该方法研究H1N1感染之后的微环境变化,想要了解呼吸道黏膜的改变是如何导致严重的继发性肺部感染。
研究方法:100位感染H1N1患者(G1)、72位感染其他流感的病人(G2)和30位健康人作为空白对照(N)。对以上样本的咽拭子进行16S扩增子测序后,利用测序数据两两比较OUT水平差异。
研究结果:三组样本分别鉴定到2788、7538和6177种OTU,其中仅有181个是共有OTU,大部分为独有的OTU。比较G1和G2组别的Shannon–Wiene指数发现,二者的OTU组成存在显著差异(p < 0.001),说明不同病毒的感染会对菌群的组成造成影响。
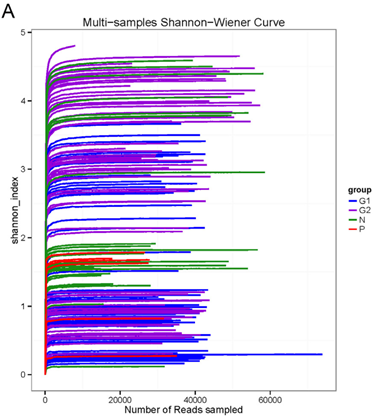
5.3 案例解析三
中文名:甲流病毒的感染影响系统微生物的动态平衡,造成肠道菌群功能缺失
英文名:Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis
发表期刊:Microbiome
影响因子:10.465(2019)
发表日期:2018
研究背景:微生物群的完整性对于生理学很重要,因此,微生物群内稳态的破坏会跟一系列病理状态有关,而共生菌可以为机体提供保护来抵抗侵入的细菌和病原。目前,对于病毒侵染宿主后的微生物群的动态构成还了解的很少。作者希望通过甲流病毒对小鼠的侵染来研究呼吸道和肠道的微生物群变化。
研究方法: 每组2个笼子,每个笼子3-6只小鼠,分别进行甲流病毒和沙门菌(S. pneumoniae)侵染。实验处理后,采集小鼠整个肺部和5 cm的小肠,提取DNA利用16s和qPCR技术进行相关检测。作者发现了躯体位点特异性和瞬时性的微生物群反应。在下呼吸道,作者发现了轻微的微生物群质量变化,在甲流感染后没有发生显著的群落数量变化。而在肠道中,甲流导致了细菌成分的减少,破坏了黏液层的完整性和潘氏细胞的抗菌多肽水平。另外,在沙门菌优势小鼠中,小肠对病原的侵染变得更加敏感。
研究结果:作者首次研究了下呼吸道和肠道在甲流病毒侵染后的微生物群的变化。利用16S高通量测序和相应的qPCR技术强调了综合数量和质量方法来准确分析物种的微生物环境。由于微生物环境的稳定性,急性感染对机体产生的失调是轻微的。但是,在一个短窗口期,某些生态位可能会失去微生物的庇护,进而容易被细菌感染。
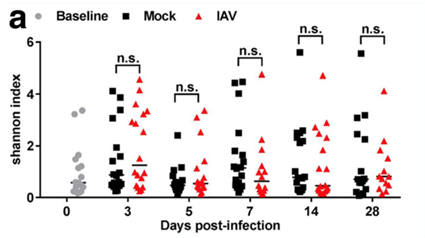
图 甲流感染对下呼吸道的微生物影响较小
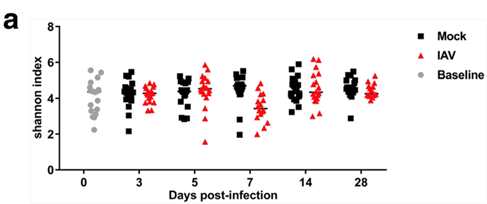
图 甲流感染降低了小肠微生物群的完整性
参考文献
[1] Vázquez-Castellanos J F, et al. Interplay between gut microbiota metabolism and inflammation in HIV infection. The ISME journal, 12.8 (2018): 1964-1976.
[2] Li Y h, et al. 16S rDNA sequencing analysis of upper respiratory tract flora in patients with influenza H1N1 virus infection. Frontiers in laboratory medicine, 1.1 (2017): 16-26.
[3] Yildiz S, et al. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome, 6.1 (2018): 9.
研究方案二
1. 研究背景
在使用不同策略治疗宿主后,宿主呈现出差异化的治疗反应,而不同治疗剂量之间也能引起差异化的治疗效果,甚至带来不同的后遗症,这可能与宿主的共生微生物组成变化息息相关。通过设置不同处理(治疗)的样本,分析不同治疗方法与治疗效果下的微生物学数据特征,阐述基本分子机制与原理。
2. 技术策略
1)扩增子测序研究不同治疗方法处理后的样本相关微生物群变化,寻找显著差异的微生物OTU。
2)宏基因组测序研究不同治疗方法处理后的样本相关微生物群变化,并深入挖掘重要菌种和功能基因。
3. 实验设计
|
分组 |
患者数 |
采样点 |
样本类型 |
样本量 |
|
治疗手段3 |
≥30 |
治疗期关键转变节点 |
粪便样本/咽拭子/口腔唾液等其他微生物样本 |
各1例/患者/时间点 |
|
治疗手段2 |
≥30 |
治疗期关键转变节点 |
||
|
治疗手段1 |
≥30 |
治疗期关键转变节点 |
||
|
对照组N |
≥30 |
安慰剂治疗 |
注:建议使用模式生物做模拟,此时可以适当减少样本量。
4. 测序策略
扩增子测序
推荐的测序平台:Illumina HiSeq2500平台
推荐的扩增区域:单V4区、双V3V4区等,如有其它需求请联系华大当地销售
推荐的测序数据量:单个样本测序数据量不低于50,000 tags
宏基因组测序
推荐的测序平台:MGISEQ平台
推荐的测序读长: PE150
推荐的测序数据量:单个样本测序数据量不低于5 Gb
5. 案例展示
中文名:益生菌处理对感染H7N9病人的影响
英文名:The Effect of Probiotic Treatment on Patients Infected with the H7N9 Influenza Virus
发表期刊:PLOS ONE
影响因子:2.776(2019)
发表日期:2016
研究背景: H7N9在2013肆虐中国东部地区。利用抗生素和益生菌进行的二次感染的预防性治疗在抗击病毒过程中变得非常重要。为了评估抗生素治疗对恢复体内平衡和降低二次感染风险的效果,作者设计了该实验项目。
研究方法: 作者收集了来自15个病人的一共113份粪便、痰和血液样本,样本分为4组,分别是无处理组a、仅抗生素处理组b、仅益生菌处理组c和抗生素益生菌双处理组d。利用变性梯度凝胶电泳鉴定其中的微生物组份。然后利用各种生物信息学工具来分析样本之间的微生物差异。同时,作者招募了20位健康人群作为对照组。
研究结果:5位病人发生了二次感染,包括肺炎克雷伯菌、鲍曼不动杆菌、白假丝酵母。在不同时间点,利用变性梯度凝胶电泳对同一个个体的粪便样本进行检测,发现结果存在明显差异,尤其是二次感染的病人。香农指数和多样性指数在感染病人中明显更低。在受益生菌处理后,没有经受抗生素治疗的病人的粪便细菌成份呈现出多样性增加的趋势。另外,作者还发现,枯草杆菌和屎肠球菌可以降低病人二次感染的风险。
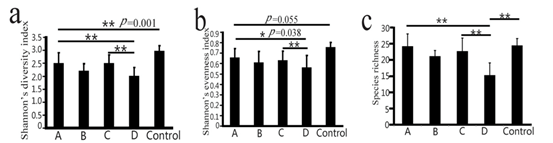
1)利用变性梯度凝胶电泳分析发现,20个健康对照组的人群的粪便微生物组成研究发现,20个样本存在较大的个体差异。
2)感染H7N9后,香农指数显示,处理组的粪便微生物多样性显著低于对照组,其中,仅抗生素处理的b组差异不显著。
参考文献
[1] Hu X J, et al. The effect of probiotic treatment on patients infected with the H7N9 influenza virus. PloS one, 11.3 (2016).
研究方案三
1. 研究背景
各地的研究者陆续在不同宿主身上发现了新的突变新冠病毒菌株,那么它们之间的关系是什么?何为先何为后?病毒突变后它的危害性和传播能力会如何变化?利用高通量测序技术,我们可以研究不同来源的病毒基因组序列,探究病毒的进化历程,为后期的诊断和治疗提供基础信息帮助。
2. 技术策略
宏基因组研究不同来源的病毒基因组序列,寻找病毒的进化和演变历程。
3. 实验设计
|
分组 |
样本数 |
采样点 |
样本类型 |
样本量 |
|
来源1 |
≥10 |
不同组织器官 |
/ |
/ |
|
来源2 |
≥10 |
不同组织器官 |
||
|
来源3 |
≥10 |
不同组织器官 |
注:其他病毒分类也可以。
4. 测序策略
宏基因组测序
推荐的测序平台:MGISEQ平台
推荐的测序读长: PE150
推荐的测序数据量:单个样本测序数据量不低于5 Gb
5. 案例展示
5.1 案例解析一
中文名:蝙蝠可能是携带新冠病毒的野生动物
英文名:A pneumonia outbreak associated with a new coronavirus of probable bat origin
发表期刊:Nature
影响因子:43.07
发表时间:2020年02月
研究背景:在过去的二十年间,冠状病毒已造成两次大规模的流行性疾病:SARS(严重急性呼吸综合征)和MERS(中东呼吸综合征)。一般认为,主要存在于蝙蝠体内的冠状病毒(SARSr-CoV)可能会导致传染性疾病的发生。2019年12月中国武汉海鲜市场爆发的不明原因肺炎病例的典型临床症状包括发热、干咳、呼吸困难、头痛和肺炎等。该疾病是由冠状病毒感染导致的,但目前冠状病毒的宿主不明。
研究方法:7名重症肺炎患者 (其中 6 人是海鲜市场的卖家或送货员)进行RNA宏基因组测序和PCR分析。
研究结果:
1)对支气管肺泡灌洗液BALF收集的样本WIV04进行宏基因组分析,过滤掉宿主来源的序列,共获得1582条clean reads,其中87.1%( 1387条reads)比对上蝙蝠体内的冠状病毒(SARSr-CoV);通过de novo组装和PCR补洞,病毒基因组全长为29 891bp,与SARS-CoV序列相似性高达79.6%,根据WHO的命名规则,命名为2019-nCoV;通过基因成分分析,主要由6个ORF和若干附属基因组成,作为冠状病毒分类的ORF1ab基因其氨基酸序列和SARS-CoV的相似度高达94.4%,说明2019-nCov和SARS-CoV同属于SARSr-CoV;共线性分析发现2019-nCoV与中菊头蝠中的Bat CoV RaTG13核苷酸一致性高达96.2%。
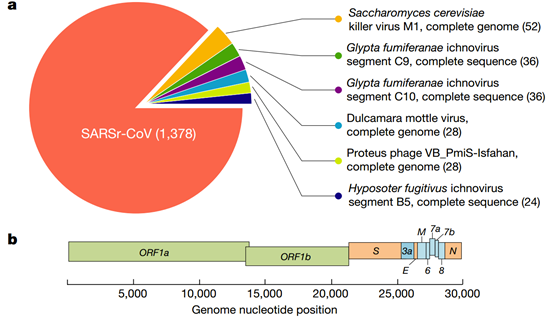
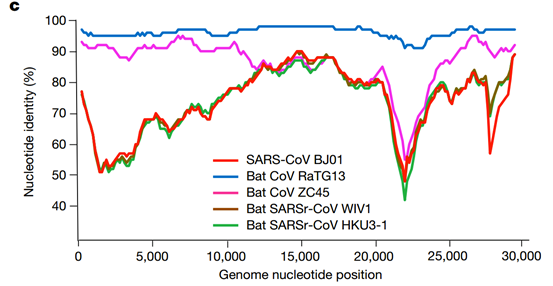
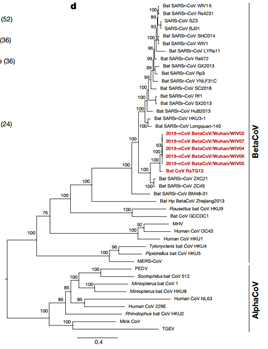
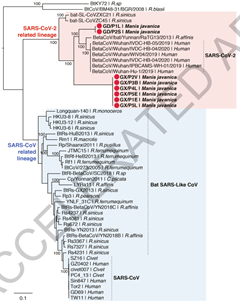
2)通过高通量测序获得其他四名患者(WIV02、WIV05、 WIV06、WIV07)的冠状病毒全基因组序列,经过比较分析发现序列间相似度高达99.9%;通过系统发育树分析发现,2019-nCoV与Bat CoV RaTG13最近缘。
5.2 案例解析二
中文名:穿山甲可能为新冠肺炎病毒的潜在中间宿主
英文名:Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins
发表期刊:Nature
影响因子:43.07
发表时间:2020年03月
研究背景:中国和世界范围内正在爆发的病毒肺炎与SARS-CoV-2冠状病毒有关。尽管蝙蝠可能是SARS-CoV-2病毒的最终宿主,但中间宿主尚不明。
研究方法:
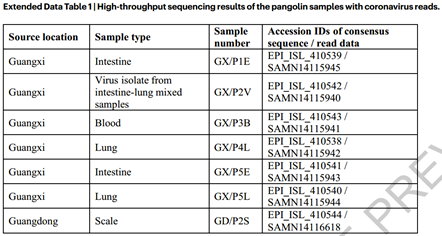
样本集1:2017年8月至2018年1月广西海关缉私行动中获得了18只马来穿山甲的43个组织(包括肺、肠、血)样本,进行RNA宏基因组测序和病毒扩增子测序。
样本集2:2018年5月至7月之间广西海关收集了12只穿山甲的19个样本(9个肠组织,10个肺组织),进行病毒QPCR检测。
样本集3:2019年3月广东海关反走私行动中查获的5份穿山甲样品(2份皮肤组织,1份未知组织,1份鳞片),进行RNA宏基因组测序。
研究结果:
1)RNA宏基因组测序后发现样本集1的43个样本中有6个样本(2个肺、2个肠、1个肺肠混合物、1个血液样本)中存在冠状病毒,结合病毒扩增子测序补洞共获得6个完整或近完整的基因组序列(GX/P1E、GX/P2V、GX/P3B、GX/P4L、GX/P5E、GX/P5L);通过构建进化树发现6个样本所含的病毒都属于新冠病毒的近缘;对GX/P2V样本中的病毒(Pangolin CoV GX/P2V)进行结构分析,发现基因组结构与新冠病毒相似度高达99.83~99.92%,具有9个开放阅读框。
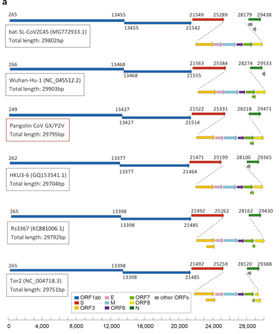
2)基于新的病毒基因组序列,设计qPCR检测引物,发现样本集2的19个样本中有3个肺组织样本呈冠状病毒阳性。
3)通过高通量测序,发现样本集3的鳞片样品中包含冠状病毒序列,并组装出1个全长为21, 505bp的部分基因组序列(GD/P2S),代表新冠病毒SARS-CoV-2基因组的72%。
4)2019年在广东进行的另一项关于患病穿山甲的研究,从肺样本中发现了与新冠病毒相似的病毒重叠群。通过不同的组装方法和人工筛选,获得了约占全长病毒基因组86.3%的部分基因组序列(GD/P1L)。
5)对β冠状病毒属的Sarbecovirus亚型的53个病毒进行系统发育树构建,发现穿山甲中发现的8个冠状病毒基因组,与SARS-CoV-2新冠病毒基因组相似率在85.5%-92.4%,并在系统进化树中代表了新冠病毒相关病毒的两个亚型。
6)由于Sarbecovirus亚属的冠状病毒成员存在广泛的基因重组,通过重组分析发现穿山甲样本的冠状病毒的受体结合域(图中红色箭头方向)和SARS-CoV-2新冠病毒受体结合域的相似性高达97.4%,而在蝙蝠冠状病毒的相似性为89.2%。
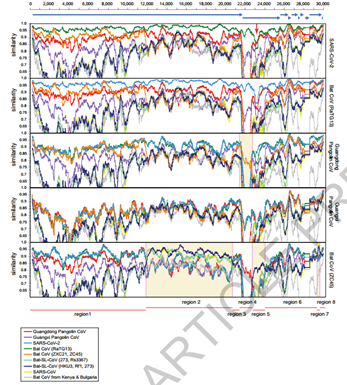
7)广东穿山甲冠状病毒和SARS-CoV-2新冠病毒在受体结合域的5个关键残基上拥有相同的氨基酸,而蝙蝠冠状病毒RaTG13和SARS-CoV-2新冠病毒只有一个氨基酸相同。然而,只针对受体结合域的同义位点系统发育分析显示,广东穿山甲冠状病毒并非SARS-CoV-2新冠病毒的最接近亲缘关系。

参考文献
[1] Zhou P, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 2020.
[2] Lam T T Y, et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature, 2020