王者变青铜,攻克超大基因组正当时!
10天完成25G的基因组组装,超大基因组迎来新突破!日前,华大基因与合作伙伴对一种裸子植物进行了测序,该物种基因组大小为25G,获得了2.2T的PacBio CLR 数据,并在10天内完成了组装。
超大基因组,一般指基因组大于10G的物种,这些物种的测序和分析工作量都非常大,尤其对于组装分析来说,是个巨大的挑战!华大基因和合作伙伴是如何在短短10天内完成了这个突破呢?
25G,只需10天
先来看看该物种的组装结果:
表1 华大在线裸子植物组装结果
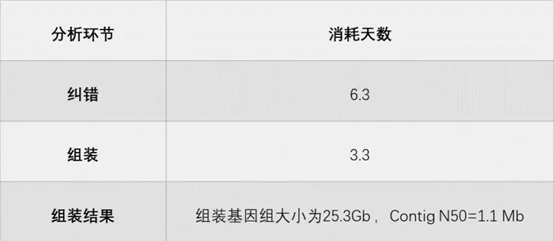
划重点!该裸子植物用的是CLR数据,纠错用的是Canu。
是不是觉得很惊奇?Canu软件以超级消耗分析资源著称,处理超大数据量时往往难以hold住。但这次足足2.2 Tb的CLR数据,用Canu 纠错居然没爆掉,而且仅仅6天多就纠错完毕。这里面的“秘密武器”在于,华大的组装人员对Canu进行了优化!
在这里,要展开说一下。前几天,PacBio 公司公布了利用HIFI数据组装另一种裸子植物——六倍体的加州红杉(基因组大小27Gb)的成果,组装结果大小为47.7Gb,Contig N50 达到1.92Mb。作为一个超大基因组,还是六倍体,这个组装结果让人惊喜。
表2 PacBio 公司公布的加州红杉的组装结果
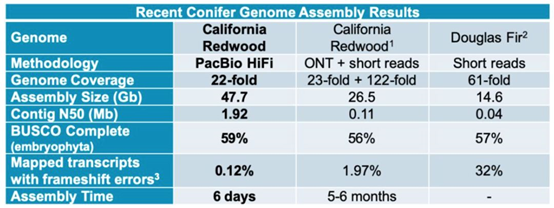
这次组装的关键,在于用了HIFI测序数据。HIFI数据本身的测序准确率就非常高(可达99%),不但让后续的组装结果更准,还大大降低纠错环节的计算资源消耗,使组装更简单。这种又快又准的优势对超大基因组,尤其是多倍体基因组的组装就会特别明显。但是,HIFI数据也有缺点,那就是由于数据产量减少,测序成本也比常规CLR数据高了数倍。
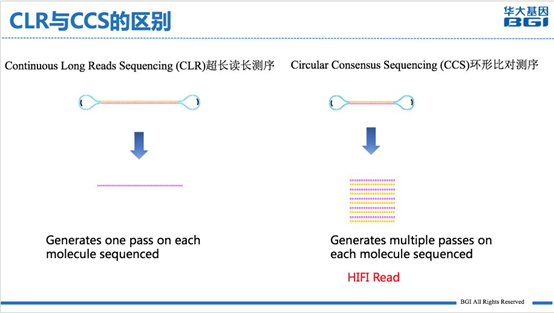
图1 PacBio HIFI数据和 PacBio CLR 数据的区别
从上图可以看到,CLR测序模式产生的数据就是基于单循环测序的结果,一个插入片段只测序一次,准确率和PacBio 常规测序保持一致,在85%左右;而HIFI测序模式(即CCS测序模式)是一个插入片段循环测序多次,下机数据可以进行自我纠错,准确率可以达到99%。
表3 CLR及HIFI测序模式的读长及测序指标
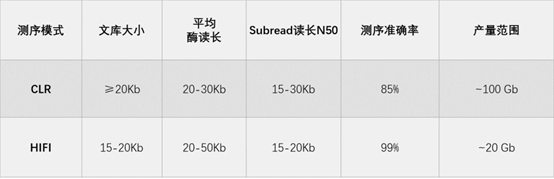
有没有使用常规CLR数据来解决超大基因组组装的方案呢?特别是对于已经测了CLR数据的用户来说,毕竟重新测一批HIFI数据则意味着要多付出好几倍的测序费用。
答案是肯定的。
CLR数据的缺点是错误率偏高。因此,组装的难点就在对CLR数据的纠错——如何在短时间内完成精准纠错。华大结合多年来积累的组装经验,对计算集群的使用效率、纠错软件等方面做了一系列的优化。得出的指标与加州红杉的效果接近,但成本却要低的多。可以说,对于二倍体的超大基因组组装,这是一套性价比很高的解决方案。
可以说,华大基因此次与合作伙伴在CLR数据方面的突破,为研究人员带来了好消息。
组装经验丰富
华大基因在超大基因组的组装上积累了丰富的经验,除裸子植物之外,还涉及超大基因组的作物、石蒜科观赏植物、八倍体水生生物、超大海洋生物等物种,数据类型涉及PB CLR/PB HIFI/ONT,组装结果Contig N50超过1Mb。
如果您有超大基因组物种,或者搞不定组装的数据,请毫不犹豫地选择华大基因,优秀的组装分析咖在等您!
拓展阅读
从王者到青铜,超大基因组组装背后的故事
作为超大基因组的典型代表——裸子植物,以基因组超大、重复序列超多、杂合度超高为“主要技能”,有时还兼具多倍体的“超级技能”,成为基因组组装界令人闻之色变的大boss。攻克难度之大,曾经让组装大咖也望而却步。能取得成绩的“强攻者”更是屈指可数——
2013年,Nature
早在2013年5月,华大参与测序的首个裸子植物——挪威云杉基因组公布并荣登Nature 杂志。研究者采用流式细胞仪预估基因组大小19.6Gb,测序采用WGS+Fosmid的策略,取样来源有单倍体和二倍体样本,单倍体测序深度 38X+二倍体测序深度55X,组装出的基因组草图大小为12Gb, scaffold N50为4.8Kb。这个组装结果如今看来比较粗糙,但以当时的技术条件,已是一大创举。
2014年,Genetics
2014年3月,另一个裸子植物——火炬松基因组发表在Genetics上,通过Kmer分析预估的基因组大小为20.4Gb。该研究通过64X短片段文库+13X Mate pair大片段文库的测序策略,获得的V1.01版本的参考序列基因组大小为23Gb,Contig N50=8.2 kb;Scaffold N50=66.9 kb。虽然基因组的组装总长度与实际大小比较接近,但是组装结果的连续性还是不高,片段比较零碎。
2016年,GigaScience
2016年11月,华大主导的裸子植物——银杏基因组发表在GigaScience上,采用短读长测序平台,测序深度196X,基因组大小10.6Gb,组装指标:Contig N50=48Kb,Scaffold N50=1.36Mb。Scaffold N50突破1Mb,银杏的组装结果有了巨大的提升。
好在,时代在发展,随着华大基因等业界代表和合作伙伴的不断求索,曾经“王者”级别难度的超大基因组,放在现在充其量也只能算个“青铜”。尤其是现在如此高性价比的方案也已经突破,超大基因组组装,就选择华大基因!
参考文献
1. Nystedt B, Street N R, Wetterbom A, et al. The Norway spruce genome sequence and conifer genome evolution[J]. Nature, 2013, 497(7451): 579-584.
2. Zimin A, Stevens K A, Crepeau M W, et al. Sequencing and assembly of the 22-Gb loblolly pine genome[J]. Genetics, 2014, 196(3): 875-890.
3. Guan R, Zhao Y, Zhang H, et al. Draft genome of the living fossil Ginkgo biloba[J]. Gigascience, 2016, 5(1): s13742-016-0154-1.