- 首页 > 基于10x Genomics Chromium X单细胞RNA-Seq
基于10x Genomics Chromium X单细胞RNA-Seq
产品介绍
2021年,10x Genomics Chromium X仪器正式推出,可以实现百万单细胞分选,核心技术原理同Controller仪器,均是基于液滴法原理,通过控制微流体的进入,将带有barcode、UMI(Unique Molecular Index,分子标签)、引物的凝胶珠(Gel Beads)与单细胞和酶混合,从而实现多种通量的单细胞分离,该仪器可以实现每次运行中分析数十万个细胞,每张芯片最多可分析16个样本,且可通过样本多重分析进一步提高通量,后续进一步建库,并结合华大DNBSEQ™高通量测序平台,实现单细胞基因表达研究,细胞类型解析及细胞异质化研究等。

图1 X仪器与配套芯片
产品亮点
1. 百万级单细胞捕获
检测通量大幅提升,一个通道最多捕获6万个细胞,一张芯片最多捕获96万个细胞,细胞分群更准确更清晰;可以鉴定到更多罕见细胞;
2. 高性价比检测
样本越多越优惠,以更低成本完成更多单细胞检测;
3. pooling模式灵活
根据实验需求,可实现至多12个样本pooling,pooling方式可以根据研究需求调整;
4. 批次效应降低
一张芯片可以实现多个样本的同时分选,减少不同批次实验产生的误差。
技术原理
- 单细胞分离原理
基于10x Genomics平台的高通量单细胞RNA-Seq技术是利用液滴法的原理,使用GemCode技术,通过控制微流体的进入,将带有Barcode(350万个,标记不同单细胞)、UMI(Unique Molecular Index,分子标签)、引物的凝胶珠(Gel Beads)与单细胞、酶混合,形成一个GEM(Gel in Emulsion)的液滴结构,实现单细胞的分离。10x Genomics X平台的液滴封装大约有65%的捕获效率,可在18分钟内完成。
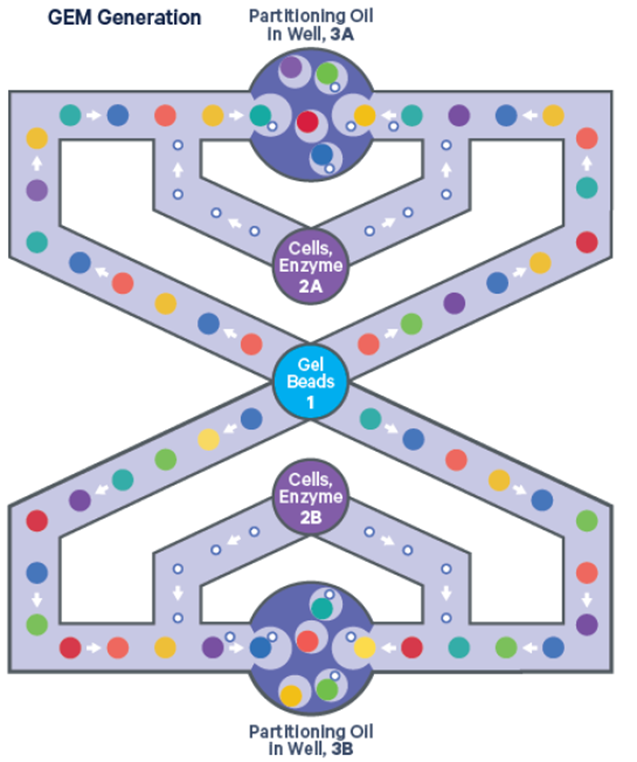
图2 10x Genomics单细胞分离原理示意图
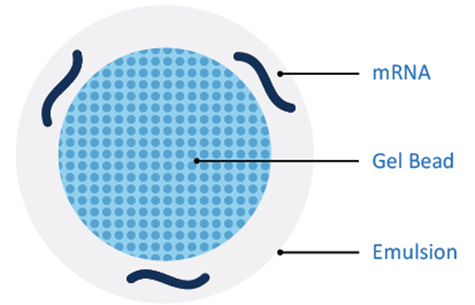
图3 GEM(10x Gel in Emulsion)结构示意图
- Pooling原理
在细胞悬液制备好之后,用样本标签标记细胞。样本标签结构示意图如下,主要包括Read2、Feature Barcode和Capture Seq2;Read2:保守序列,用于后期扩增;Feature Barcode:12种序列,区分不同样本;Capture Seq2:与引物序列互补,用于反转录样本标签。样本标签经建库测序后,通过读取样本标签上的Feature Barcode序列,可以区分转录本来自于哪一个样本。
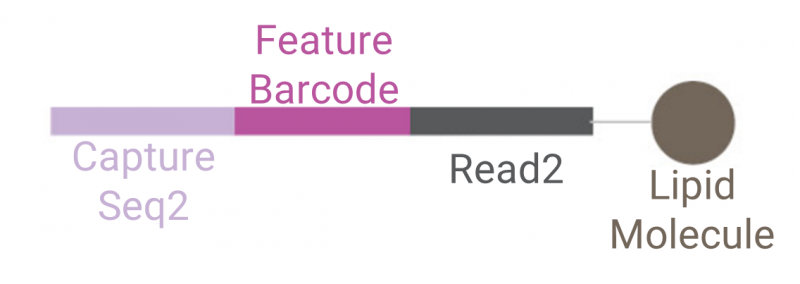
图4 样本标签结构示意图
实验流程

产品类型
- Pooling版- 多样本标准通量单细胞分选模式; 预计产出8000-10000个单细胞/样本(同一物种细胞,前期限人、小鼠);
- 非pooling版- 高通量单细胞分选模式;20000个单细胞/样本
*以上版本均包含建库测序Dr. Tom标准分析
测序策略
推荐DNBSEQ平台测序
测序策略: PE100
测序深度:推荐平均20-50k reads/cell
预计产出数据量(数据产出波动上下20%左右):
- pooling模式:8000-10000细胞,~650M reads,2条lane/样本
- 非pooling模式:20000细胞,~1300M reads,4条lane/样本
信息分析流程
测序得到的原始数据我们称之为Raw reads,之后我们会根据X平台单细胞RNA-Seq的文库结构,对Reads的10x Barcode、 UMI 和插入片段部分进行拆分,之后插入片段部分将比对到参考基因组,然后统计比对到各个区域的比例,并进行表达量统计;基于表达量的结果,进行细胞时间轨迹预测,细胞聚类等分析,基于差异表达的结果,进行KEGG富集,GO富集,蛋白互作网络分析等。
表1 信息分析内容
|
信息分析条款 |
信息分析内容 |
|
标准信息分析 (需要基于良好的参考序列, 且每个样本拆分出的细胞数目上下浮动在20%,或以内的误差皆属于正常范围) |
1. 测序结果统计 2. 数据质控统计 3. 比对结果统计 4. 基因表达定量分析 5. Marker基因鉴定 6. 细胞类型注释 7. 单样本/多样本细胞聚类分析 8. 特异marker基因差异分析 9. 样本间相同cluster基因差异分析 10. cluster差异表达基因GO功能分析 11. cluster差异表达基因Pathway功能分析 12. cluster差异表达基因TF编码能力预测 13. cluster差异表达基因蛋白互作分析 14. cluster基因相关性网络分析 |
|
高级信息分析 |
细胞轨迹分析(仅选取时间点样本可选做) |
|
定制化信息分析 |
可结合客户的需求,协商确定定制化信息分析内容。 |
案例一:人类全面胚胎基因表达图谱

英文标题:A human cell atlas of fetal gene expression
发表期刊:Science
影响因子:47.720
发表时间:2020.11
样本类型:72天到129天不等的人类胚胎器官组织样本,共400万个单细胞
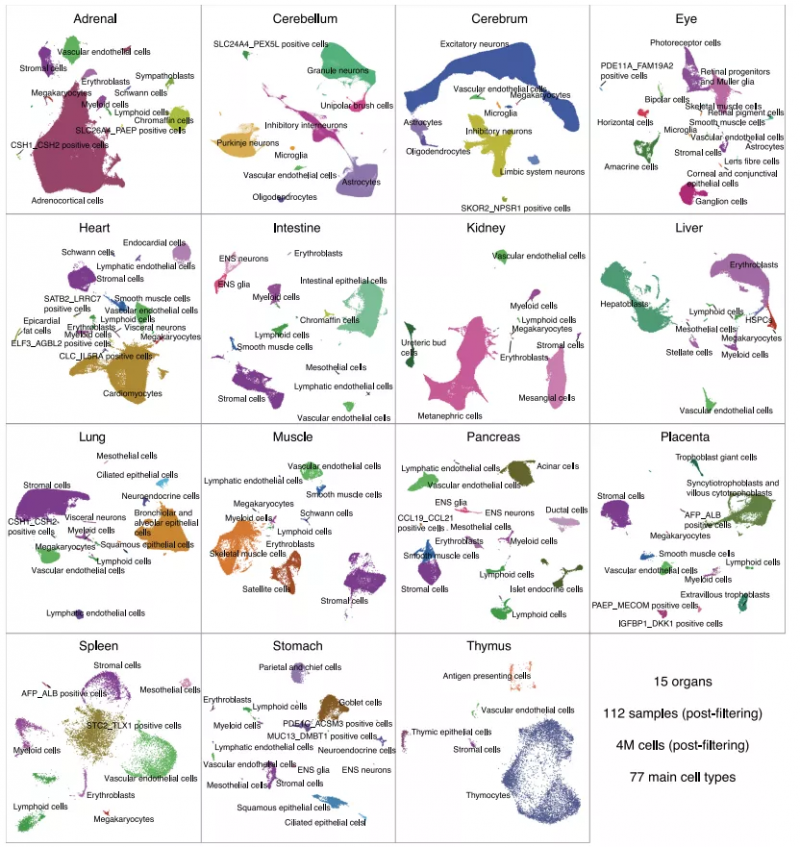
图1 人类胚胎器官的细胞图谱
研究成果:
1. 构建了胎儿组织的基因表达细胞图谱。
2. 在多个器官中具有代表性的细胞类型,以及与发育相关的细胞类型趋向于共定位。在77种主要细胞类型中,许多表面蛋白、分泌蛋白、转录因子和非编码RNA表达差异显著。
3. 尽管物种不同,但许多人类细胞亚型与小鼠细胞类型匹配达到了1:1。并针对主要细胞类型(包括血液、内皮细胞和上皮细胞)进行了跨组织综合解析。
4. 鉴定识别并验证了组织中潜在循环的滋养细胞样细胞和肝母细胞样细胞。
5. 对于血细胞,该研究绘制了从造血干细胞到所有主要亚系细胞状态轨迹的多器官图谱。
研究意义:为单细胞受精卵开始的人类细胞类型多样性的研究提供支持。有助于跨越不同细胞类型探索人体基因表达,更好地解析细胞类型及其发育轨迹。
案例二:15种肿瘤的单细胞转录组图谱

英文标题:A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells
发表期刊:Cell
影响因子:41.583
发表时间:2021.02
样本类型:15个癌种内近14万个肿瘤浸润的髓系细胞
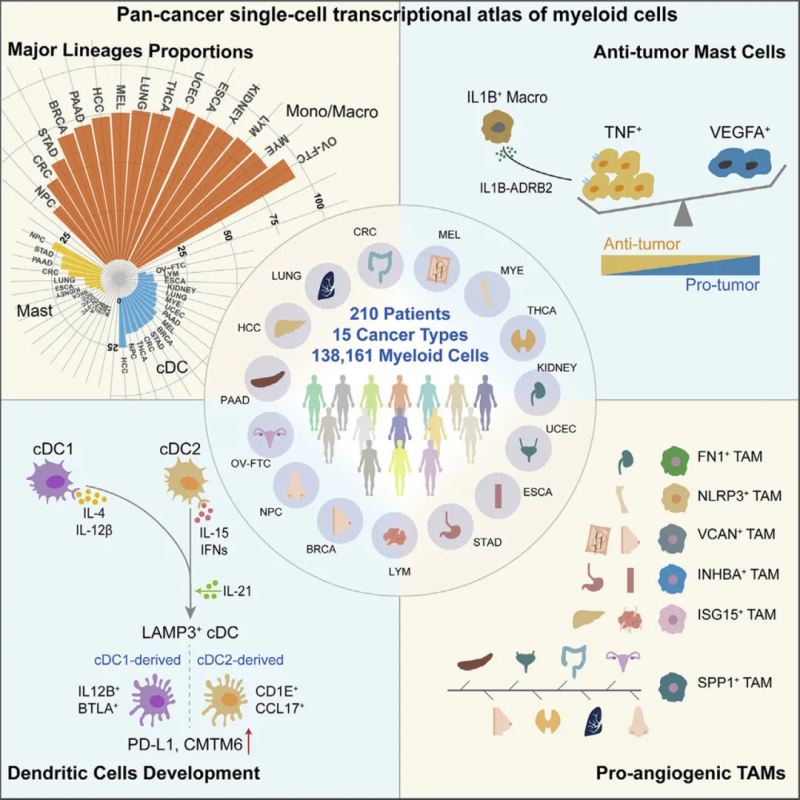
图2 肿瘤浸润的髓系细胞转录图谱
研究成果:
1. 刻画出肿瘤浸润髓系细胞类群在不同癌种内的特征图谱。
2. 虽然肿瘤相关巨噬细胞在不同癌种内表现出较高的异质性,但不同类型的肿瘤中均存在一群具有促血管生成功能的巨噬细胞类群。
3. 具有促血管生成功能的细胞类群的特征标记基因不尽相同,但其转录组具有较高的相似性,而且这个细胞类群与患者的预后具有显著的负相关。
研究意义:首个泛癌种尺度下的单细胞肿瘤免疫图谱研究。揭示了肿瘤微环境的复杂性和多样性,为未来开发新的免疫治疗策略提供了关键的线索和潜在的靶点。
案例三:癫痫影响神经元的基因表达

英文标题:Identification of epilepsy-associatedneuronal subtypes and gene expression underlying epileptogenesis.
发表期刊:Nature Communications
影响因子:14.913
发表时间:2020.10
样本类型:多个颞叶癫痫和非癫痫患者颞皮层样本中的11万个神经元

图3 癫痫和非癫痫患者颞叶皮层的单核转录组
研究成果:
1. 确定了癫痫皮层中多种神经元亚型的大规模转录组变化,并找到了受癫痫影响最大的确切神经元。
2. 最大的转录组变化发生在几个主要神经元家族(L5-6_Fezf2和L2-3_Cux2)和GABA能中间神经元(Sst和Pvalb)的不同神经元亚型中,而同一家族中的其他亚型则受影响较小。
3. 谷氨酸信号传导是癫痫病失调最严重的信号之一。
研究意义:这项研究是第一个探索人脑癫痫区中每个神经元如何受到癫痫影响的研究,揭示了癫痫中基因表达的复杂性,确定了最有可能促成癫痫发生的表达变化的基因,为治疗癫痫提供了理论基础。
参考文献:
[1] Cao J, O'Day DR, et al. A human cell atlas of fetal gene expression. Science. 2020 Nov 13;370(6518):eaba7721. doi: 10.1126/science.aba7721. PMID: 33184181; PMCID: PMC7780123.
[2] Cheng S, Li Z, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021 Feb 4;184(3):792-809.e23. doi: 10.1016/j.cell.2021.01.010. PMID: 33545035.
[3] Pfisterer U, Petukhov V, et al. Identification of epilepsy-associated neuronal subtypes and gene expression underlying epileptogenesis. Nat Commun. 2020 Oct 7;11(1):5038. doi: 10.1038/s41467-020-18752-7. PMID: 33028830.
结果展示
样本要求 | 类型 | 组织:新鲜/冻存的肿瘤组织,新鲜人全血 |
细胞:新鲜/冻存肿瘤细胞、生殖细胞、胚胎细胞、免疫细胞、PBMC、其他原代细胞、细胞系等。Pooling模式要求同一物种细胞。 | ||
细胞状态 | 细胞直径<30μm;细胞活性≥80%;细胞数>5×105/样本 | |
细胞悬液背景干净,无大量结团、碎片及杂质,不含Ca2+和Mg2+ |
交付周期
从样品复苏后,镜检符合上机要求、预付款到位开始(不包括由于样品等问题停滞时间以及试剂订购周期),细胞类型及生物信息分析整体完成周期视不同的类型而有所变化。
DNBSEQ平台
- 样品数≤8个,45个工作日(含建库、测序、标准信息分析);
- 样品数≥9个,请单独咨询周期。
Q1:为什么一个通道在pooling模式下可上4万多个细胞,非pooling模式下只能上2万个细胞?
当细胞上样量过大时,会出现较高的双胞率。非pooling模式下,双胞无法排除。pooling模式下,双胞包括相同样本和不同样本的双胞。相同样本的双胞不能排除。不同样本的双胞里含有不同的样本标签,因此可以舍弃。舍弃后,整体的双胞率(相同样本的双胞)在可以接受的范围内。所以pooling模式下,可以实现更高的细胞通量。
Q2:Pooling样本有什么要求吗?
限同一物种的细胞。
Q3:X平台是如何实现多个样本混合建库的?后续又如何判断转录本的样本来源?
上样之前,将各个样本分别与不同的样本标签(12种可选择)混合。标签含有脂质分子,可与细胞膜结合。不同标签的Feature Barcode序列不一样,以此来标记不同的样本。
凝胶珠上覆有相同10x Barcode序列的引物,其中包括转录组引物和样本标签引物,分别与转录组mRNA和样本标签结合,随后各自反转录建库。这两种文库的10X Barcode序列相同,因此可以区分转录组来源于哪一个样本。
Q4:样本pooling模式下,每个样本细胞产出均一吗?
细胞捕获效果与细胞计数和质量有关系。细胞计数要准确,不同样本保证相同上样量。细胞质量要好,活率相近。
Q5:通过X平台进行单细胞RNA-Seq的组织样品,是否可以提供前期处理的方法?
目前单细胞送样建议手册上有提供通用的组织消化方法,如下可供参考。
需要注意,不同组织存在组织及细胞的特异性,使用通用组织消化方法的效率会因组织类型差异,存在消化效率的差异。
Q6:X平台单细胞RNA-Seq采用的3’端扩增技术与Smart-seq2的扩增技术有什么区别?
Smart-seq2扩增技术针对是的3’~5’端全部的mRNA;3’端扩增技术主要是扩增3’端,是否能延伸至5’端,取决于酶的活性等系列实验因素决定。因此,扩增产物的长度上会有一定的区别和差异。
Q7:如果客户现场分离了1000个细胞,并检测出细胞活度很高,问:可否直接使用1000个细胞进行X平台上机?
不建议。因为X平台单细胞RNA-Seq最低建议细胞送样量为5*105个细胞,且活率建议在80%以上。
Q8:细胞物种没有相应的参考基因组,可以做这个产品吗?
产品分析需要基于一个良好注释和组装的参考序列。基因注释不完善或只注释到转录本,没有注释到基因等的不完善参考序列,会出现占用内存过多,即使对转录本进行去冗余及重新构建,跑出的结果可能会不太好,需要客户明确其风险因素。