- 首页 > 人全基因组重测序
人全基因组重测序
DNBSEQ人全基因组重测序(WGS),采用拥有自主知识产权的测序仪和云计算平台,为广大科研工作者提供高准确度、高性价比的基因组测序服务和一站式科研解决方案,支持大型队列研究,助力精准医学。
DNBSEQ测序平台
华大基因DNBSEQ测序平台采用的是DNB(DNA Nanoball ,DNA纳米球)[1]核心测序技术,独特的线性扩增模式。
DNB技术是目前全球少有的能够在溶液中完成模板扩增的技术,能够在扩增过程避免错误累积的发生,有效提高测序准确度。因为是基于滚环扩增,DNB技术不仅有效增加了待测DNA的拷贝数,大大增强了信号强度,且同一个模板进行滚环复制,即使复制过程中引入单个碱基的复制错误,这个错误也不会像PCR那样把这个信号放大。
完成模版扩增后,DNB将转载到Patterned Array(规则阵列)上。Patterned Array采用先进的纳米硅半导体精密加工工艺,使用率高,单位测序成本更低。DNB是在溶液里面提前扩增完成的,在loading过程中没有聚合酶、引物和dNTP等PCR条件,所以华大自主测序平台从测序原理上有效的避免了大量duplicates的产生。
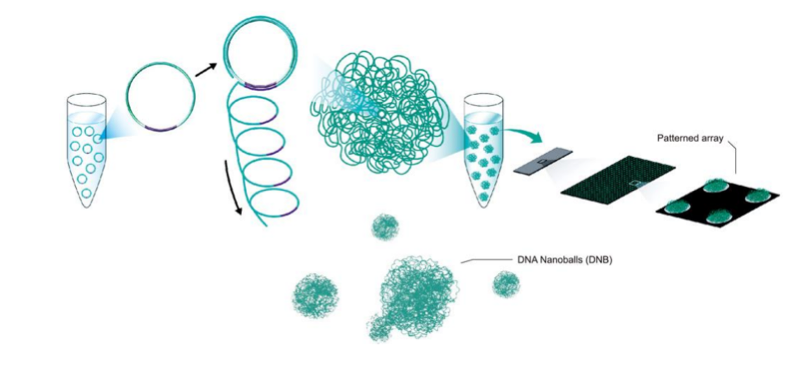
图1 DNBSEQ平台测序原理
给您选择我们的八个理由
- 稳定的产出高质量测序数据
对随机挑选的1,000+条lane DNBSEQ平台 WGS数据质量值进行统计分析,下机Raw data Q20平均值为96.16%,Raw data Q30平均值为87.86%。
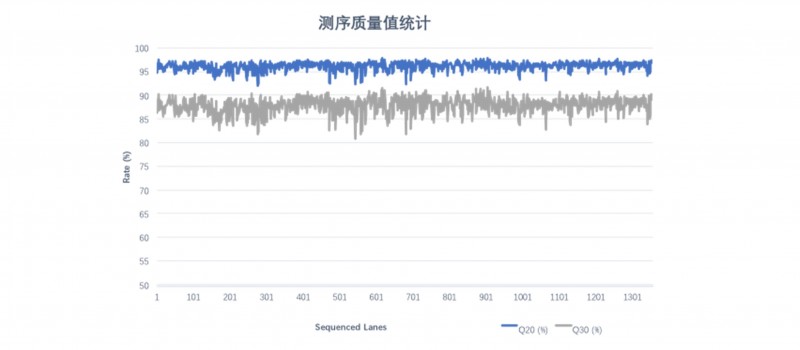
图2 1000+条lane WGS序质量统计
- 低duplicates获更多有效数据和更高覆盖度
Duplicates低,用更少的数据量,得到更多的高准确和高覆盖度的比对数据,可以发现更多变异位点,有助于挖掘疾病的低频和罕见突变,获取更加全面的基因组变异信息。
表1 主流二代测序平台标准品duplicate比率、有效测序深度及覆盖度比较
|
Sample |
X 测序平台 |
N测序平台 |
DNBSEQ平台 |
|
Raw bases (Mb) |
99998.92 |
100001.72 |
100236.61 |
|
Clean bases (Mb) |
96314.26 |
98955.15 |
99886.02 |
|
Mapping rate (%) |
99.61 |
98.68 |
99.47 |
|
Unique rate (%) |
87.18 |
86.41 |
93.31 |
|
Duplicate rate (%) |
9.65 |
10.15 |
3.02 |
|
Mismatch rate (%) |
0.8 |
0.51 |
0.48 |
|
Average sequencing depth (X) |
29.08 |
29.52 |
32.8 |
|
Coverage (%) |
99.06 |
99.06 |
99.1 |
|
Coverage at least 4X (%) |
98.57 |
98.43 |
98.62 |
|
Coverage at least 10X (%) |
97.77 |
97.2 |
97.67 |
|
Coverage at least 20X (%) |
91.8 |
89.45 |
92.97 |
- 高精准度和敏感度的变异结果
已发表文章结果显示,BGISEQ-500自主平台与HiSeq 2500测序平台变异检测的精准度(Precision)和敏感度(Sensitivity)相当[2]。
表2 BGISEQ-500与HiSeq 2500变异精准度和敏感度比较[2]
|
SNP |
BGISEQ-500 |
HiSeq 2500 |
|
Precision |
99.78% |
99.86% |
|
Sensitivity |
96.20% |
96.60% |
- 罕见突变检出率及与芯片分型的一致率高
DNBSEQ平台变异结果与Illumina Human Omni基因分型芯片评估,结果表明罕见突变检出率高,且检出的罕见突变与芯片分型结果的一致性高。
表3 DNBSEQ平台 30X rare SNP detection rate
|
Genotyping chip |
MAF |
NO. of rare SNP |
NO. of detection |
NO. of concordance |
检出率 |
一致率 |
|
OMNI |
< 2% |
7414 |
7142 |
7132 |
96.33% |
99.86% |
|
OMNI |
< 1% |
3151 |
3025 |
3018 |
96.00% |
99.77% |
|
OMNI |
< 0.5% |
1129 |
1075 |
1070 |
95.22% |
99.53% |
- 无Index hopping担忧
DNBSEQ测序仪利用独特的DNA纳米球(DNB)技术,仅使用单个index就实现了前所未有的0.0001%至0.0004%低样本错误分配率。用水代替DNA,加入index,增加空白对照,DNB测序平台发生错误匹配的概率为36 million reads分之一,即0.0000028%[3]。

图3 不同测序技术的index hopping比例
- 满足多种样本类型的需求
DNBSEQ平台WGS数据来源样本种类多样,其中包含福尔马林固定石蜡包埋(Formalin Fixed and Paraffin Embedded,FFPE)样品、单细胞样品、血液样品、基因组DNA样品、唾液样品、常规冷冻保存的新鲜组织样品等。常规基因组建库测序成功率为99%,对于降解样品如FFPE等,建库测序成功率也在90%以上。
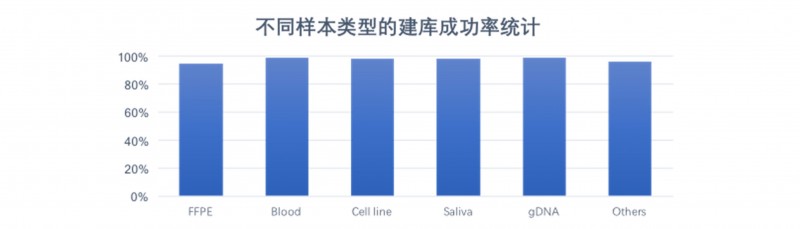
图4 DNBSEQ平台不同类型样本交付成功率
- DNBSEQ WGS PCR-free文库
PCR-free建库 + DNB (DNA纳米球)核心测序技术,为您还原真实的全基因组序列。PCR-free WGS 高质量InDel从75%提升到86%,而低质量InDel从12%降低到3%[4],PCR-free建库方法可明显提高InDel calling的精准度和敏感度。
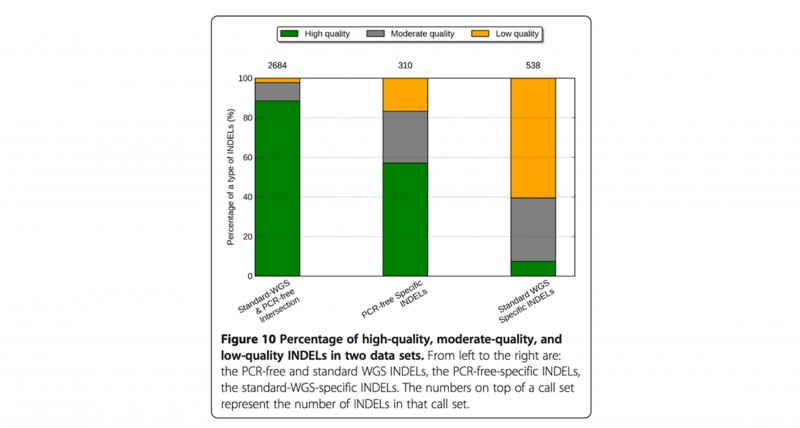
图5 高质量、中等质量和低质量InDel在不同建库方法的分布
参考文献
[1] Drmanac R, Sparks A B, Callow M J, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays.[J]. Science, 2010, 327(5961):78-81.
[2] Jie Huang, Xinming Liang, Yuankai Xuan, et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience, 2017.
[3] Li Q, Zhao X, Zhang W, et al. Reliable Multiplex Sequencing with Rare Index Mis-Assignment on DNB-Based NGS Platform. bioRxiv, 2018: 343137
[4] Han F, Wu Y, Narzisi G, et al. Reducing INDEL calling errors in whole genome and exome sequencing data[J]. Genome Medicine,6,10(2014-10-28), 2014, 6(10):89.
案例1 ChinaMAP 分析 10,588 个人的高深度全基因组序列[1]

研究材料:10588个中国人,随机选择于中国27个省份的8个民族(汉族、回族、满族、苗族、蒙古族、彝族、藏族和壮族) 。平均基线年龄为54岁,女性为64.8%。
分析内容:构建高质量的中国人群遗传变异数据、中国人群体结构分析、基因组特征比较以及变异频谱和致病性变异解析。该研究也通过全基因组关联分析探索了中国人群中2型糖尿病和肥胖遗传相关因素。在血糖相关分析中,结果验证了部分已知的2型糖尿病风险高频基因位点,包括CDKAL1、SLC30A8、SND1-PAX4、IDE-KIF11-HHEX、CDKN2A-CDKN2B、KCNQ1 等,也鉴定和发现了DENND5B、ORM1 、MAFA、PAX6、SOX4 等新位点。
重要研究成果:
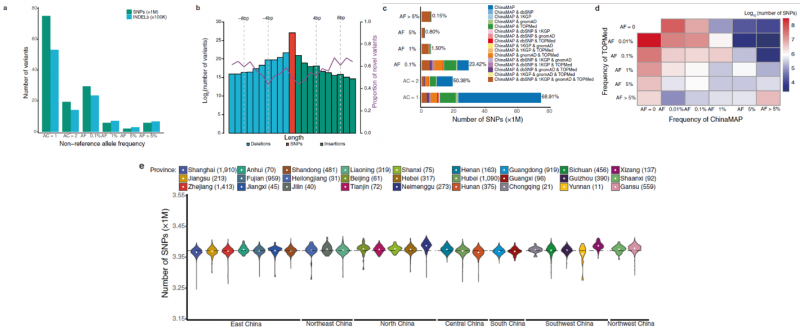
图1 ChinaMAP基因变异的分布和模式
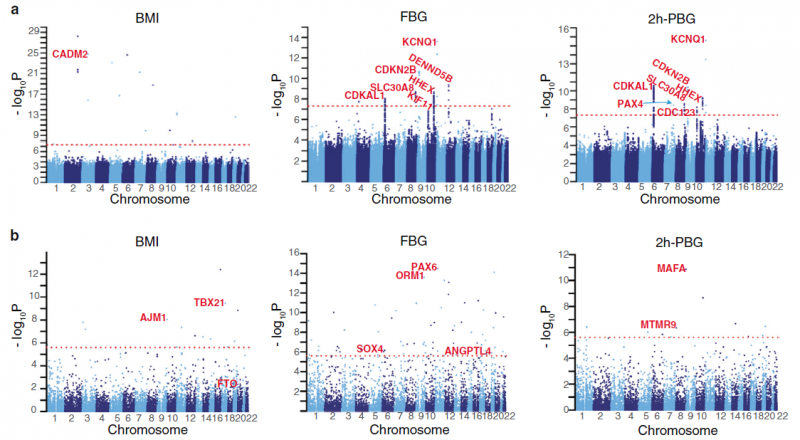
图2 体重指数与血糖的关联分析
ChinaMAP研究也通过全基因组关联分析探索了中国人群中2型糖尿病和肥胖遗传相关因素。在体重指数BMI相关分析中,研究团队发现了新的东亚人群特异性CADM2基因位点,CADM2在动物研究中已证实参与调节体重和能量稳态。而FTO等基因在欧美人群中发现的重要肥胖相关基因位点,在ChinaMAP研究结果中并不显著。从这些发现可以提示我们,对大规模中国人群特异性的基因组学的研究,对分子机制和个体化诊治的精准医学体系建立很重要。
案例2 NBT:主流高通量测序仪在人/细菌/宏基因组测序评测结果发布[2]

由生物分子资源设施协会(Association of Biomolecular Resource Facilities, ARBF)主导的ABRF NGS II期研究成果发表于Nature Biotechnology,文章题为“Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study”。研究团队基于来自Illumina、Pacific Biosciences、Thermo Fisher Scientific、BGI、Oxford Nanopore Technologies和Genapsys的多款测序平台,在多个实验室对同一人类基因组家族、三个单独菌株和十种细菌的宏基因组混合物进行测序,并将各平台数据进行全方位、系统性比较,分析各个测序平台的性能差异和测序质量,以提供真实全面的参考证据。
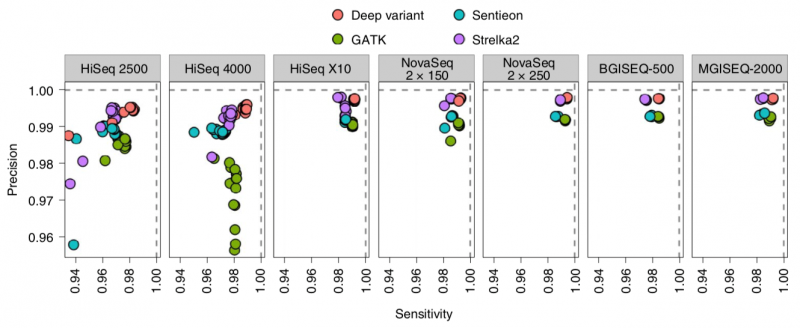
数据显示,在短读长测序平台中,DNBSEQ平台提供了非常低的测序错误率。且SNP/Indel检测的灵敏度和准确度表现也非常优秀。
案例3 DNBSEQ基因组测序揭示肺鳞癌的潜在治疗靶标[3]
Genomic sequencing and editing revealed the GRM8 signaling pathway as potential therapeutic targets of squamous cell lung cancer
肺腺癌和肺鳞癌(LUSC)是肺癌的主要病理类型,肺鳞癌占原发性肺癌的40%~51%。目前已经有多种靶向药物应用于肺腺癌,但是肺鳞癌的治疗靶点尚没有突破性进展。文章通过外显子重测序(WES),人全基因组重测序(WGS)、靶区域捕获测序(TS)和CRISPR-Cas9基因组编辑技术,利用鳞状细胞肺癌手术肿瘤和对应的源自患者的异种移植瘤(PDX)样本探索和验证肺鳞癌的潜在治疗靶标。
文章亮点:
- Illumina HiSeq X Ten平台WES测序+ DNBSEQ平台WGS测序+ DNBSEQ平台TS测序,多平台数据联合分析
- LUSC PDX模型可广泛应用于潜在治疗目标和策略的验证
- 使用CRISPR系统对PDX肿瘤细胞中的驱动基因进行功能验证
通过基因组测序和CRISPR-Cas9基因组编辑的综合分析,在手术和PDX肿瘤上整合鉴定并验证了GRM8对LUSC肿瘤的促进功能。cAMP活化剂和MEK抑制剂可显著阻断具有GRM8突变的LUSC肿瘤细胞的增殖和存活。因此,GRM8信号传导通路的组成分子可能成为携带GRM8激活突变的鳞状细胞肺癌的治疗靶标。
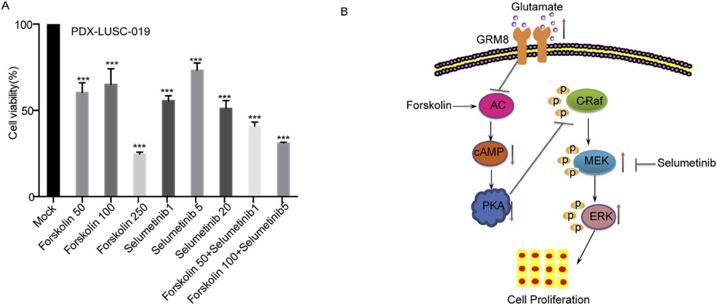
图 cAMP激活剂和MEK抑制剂作为治疗策略
图A:Forskolin和Selumetinib的联合治疗对PDX肿瘤细胞活力具有抑制作用
图B:GRM8活化通过抑制cAMP通路和激活MAPK通路促进LUSC细胞的增殖
案例4 BGISEQ-500和HiSeq X Ten全基因组测序鉴定生殖细胞和体细胞变异[4]
Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing
该研究使用BGISEQ-500平台对三种恶性胸膜间皮瘤及其对照的正常样本进行全基因组测序,并与Illumina HiSeq X Ten平台测序结果进行评估。两平台数据均使用相同的分析流程,分别比较生殖细胞和体细胞单核苷酸变异(SNP)、小插入或缺失(InDel)。结果表明BGISEQ-500平台通过全基因组测序来鉴定肿瘤样本的体细胞和生殖细胞突变是有潜力的可适用性的平台,这也是该平台首次公开可用的癌症基因组数据。
研究结果:
- 生殖细胞突变:
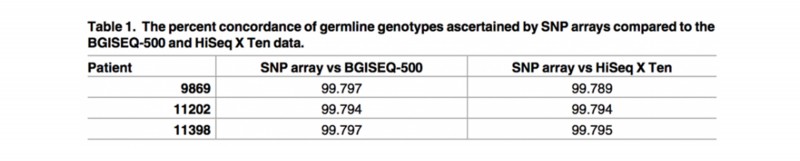
结果显示,BGISEQ-500平台和HiSeq X Ten平台识别SNP的能力与SNP分型芯片(Infinium Omni2.5–8, Illumina )是高度一致的(> 99%)。在两个测序平台中鉴定的生殖细胞SNV和indels也是高度一致(分别为86%和81.5%)。
表 SNP芯片数据分别与BGISEQ-500和HiSeq X Ten数据比较,生殖细胞突变基因型一致性的百分比
- 体细胞突变:
三名患者中总共10,890个体细胞SNV,大部分体细胞SNV(72%)在两个平台中被识别,小部分为BGISEQ-500和HiSeq X Ten两平台特有的(分别为14%,14%)。
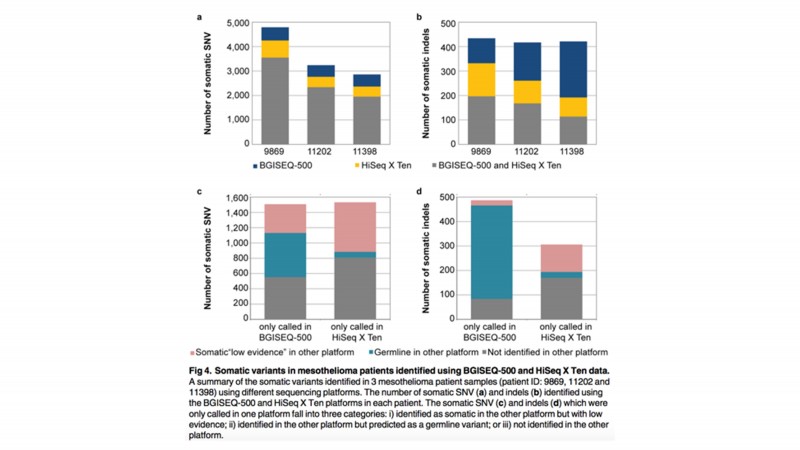
图 利用BGISEQ-500和HiSeq X Ten的数据鉴定3个胸膜间皮瘤的体细胞突变
案例5 基于DNB的测序平台可有效避免index hopping[5]
Reliable Multiplex Sequencing with Rare Index Mis-Assignment on DNB-Based NGS Platform
本研究使用三种主要的文库制备方法研究了DNB测序平台的Index hopping问题。DNBSEQ测序仪利用独特的DNA纳米球(DNB)技术,基于滚环复制(RCR)进行文库扩增,这种线性扩增可以避免常规PCR带来的错误累积。基于DNB的NGS应用仅使用单个index就实现了前所未有的0.0001%至0.0004%低样本错误分配率。此外,用水代替DNA,加入index,增加空白对照,DNB测序平台发生错误匹配的概率为36 million reads分之一,即0.0000028%。

图 不同测序技术的index hopping比例
研究结果:
- DNA纳米球技术的高index保真度
DNBSEQ平台将DNB加载到规则阵列(patterned arrays)上,并利用组合引物锚定测序技术(cPAS)进行测序。 独特的DNB技术采用具有强链置换活性的Phi29聚合酶和能够进行线性扩增的RCR工艺,每个扩增循环都以原始的单链环状DNA文库为模板,保持每个拷贝子的独立性(图1a)。因此,即使出现寡核苷酸的index hopping等错误,也不会累积错误拷贝,正确的序列总是会在后面的DNA拷贝中复制,保证高的扩增保真度。
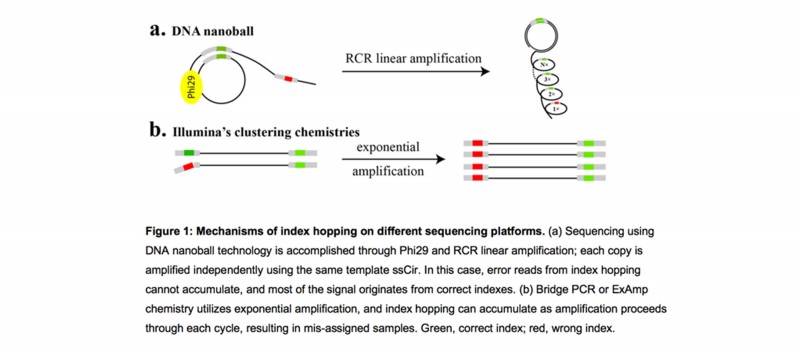
图 Index hopping在不同的测序平台产生的机制
- PCR-free文库index hopping污染率极低
除了常规PCR文库外,文中还对PCR-free文库在DNBSEQ平台的index hopping情况进行调查,未经过任何Q30过滤的99.9998%精度再次证实了DNB可以在很大程度上降低index污染。与上面的常规PCR文库类似,污染率平均约为0.0004%。
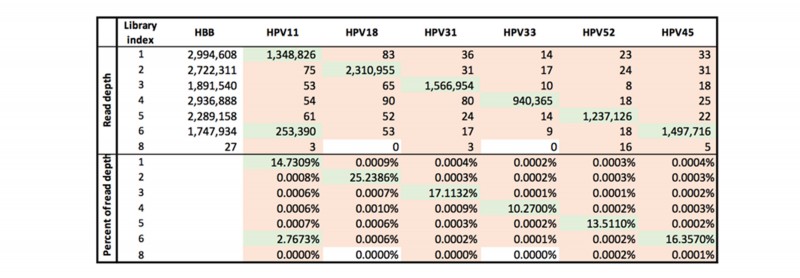
表 PCR-free 文库index污染比率
研究意义:
1、 高的检测准确度,保证体细胞低频突变、HPV检测等基因检测的准确性;
2、 Single index避免了繁琐的non-combinatorial dual index带来的额外成本和劳动力浪费;
3、 避免大通量测序中样本数据完整性的丢失。
参考文献
[1] Cao Y, Li L, Xu M, et al. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals[J]. Cell research, 2020, 30(9): 717-731.
[2] Foox, J., Tighe, S.W., Nicolet, C.M. et al. Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study. Nat Biotechnol 39, 1129–1140 (2021).
[1] Genomic sequencing and editing revealed the GRM8 signaling pathway as potential therapeutic targets of squamous cell lung cancer.[J]. Cancer letters, 2018.
[2] Patch A M, Nones K, Kazakoff S H, et al. Germline and somatic variant identification using BGISEQ-500 and HiSeq X Ten whole genome sequencing.[J]. Plos One, 2018, 13(1):e0190264.
[3] Li Q, Zhao X, Zhang W, et al. Reliable Multiplex Sequencing with Rare Index Mis-Assignment on DNB-Based NGS Platform. bioRxiv, 2018: 343137
1、标准品数据展示
测试样本选用了“瓶中基因组(Genome in a Bottle)”的人类样本NA12878,这是目前被世界上认为研究相对透彻的二倍体人类基因组,并发布了高置信变异集,可作为一个重要工具来了解测序仪和检测结果的表现。X测序平台的数据为I 公司官网下载的数据,并且,两个平台的分析均严格采用了GATK Best Practices推荐的流程进行分析。
- 高测序数据质量
测试数据有至少97%的碱基识别准确率高达99%,至少89%的碱基识别准确率高达99.9%。
Q20>97%,Q30>89%
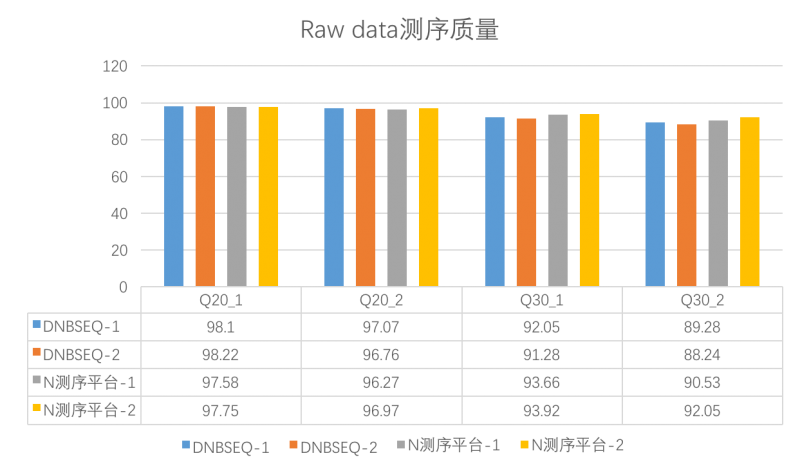
图 Raw data测序质量
测序质量值可衡量碱基未正确检出的概率。测序技术,一种类phred算法[1,2]会为片段中的每个碱基分配一个质量分值,与最初因桑格测序实验而开发的算法类似。
一个给定碱基的测序质量分值Q定义为下面的等式: Q = -10log10(e) 其中,e为预计碱基检出不正确的概率。如下所示,质量分值20表示错误率为1/100,相应的检出精确度为99%。
表 标测序质量分值与碱基检出精确度的关系

- 高比对率和覆盖度
分别在DNBSEQ和N两个平台上测了90Gb、97Gb数据和105Gb、111Gb数据。从下表可以看出,由于N平台Duplicate rate较高,需多测10%的数据,才有和DNBSEQ平台相当的有效深度。
表 DNBSEQ与N平台比对数据比较
|
Sample |
DNBSEQ-1 |
DNBSEQ-2 |
N平台-1 |
N平台-2 |
|
Clean bases (Gb) |
90.5 |
97.2 |
105.4 |
111.8 |
|
Mapping rate (%) |
99.96 |
99.89 |
99.88 |
99.88 |
|
Unique rate (%) |
93.88 |
92.53 |
82.17 |
76.88 |
|
Duplicate rate (%) |
1.84 |
3.62 |
14.83 |
20.18 |
|
Mismatch rate (%) |
0.29 |
0.3 |
0.35 |
0.31 |
|
Average sequencing depth (X) |
29.65 |
31.3 |
29.75 |
29.55 |
|
Coverage (%) |
99.2 |
99.2 |
99.14 |
99.1 |
|
Coverage at least 4X (%) |
99.02 |
99.04 |
99 |
98.92 |
|
Coverage at least 10X (%) |
98.5 |
98.69 |
98.68 |
98.49 |
|
Coverage at least 20X (%) |
91.07 |
94.44 |
92.74 |
91.97 |
|
Coverage at least 20X (%) normalized |
91.5 |
93.17 |
93.02 |
92.49 |
Clean bases:过滤掉接头,低质量和含N的reads后剩下的碱基数量;
Mapping rate:碱基比对率,比对到参考基因组的碱基数目除以clean data的碱基数目,如果测序样本存在污染或者与参考基因组差异较大,比对率偏低会影响后续的信息分析;
Unique rate:比对到基因组上唯一位置的base比率,一条reads在相同数量的容错时会有两个或者两个以上的位点都吻合,那么,它的比对结果不唯一。对于某些下游分析,需要去除比对多个位点的reads,只保留唯一比对的reads;
Duplicate reads:重复的 reads 所占比例,为了保证后续变异分析的准确性,会去掉duplicate reads后进行下游信息分析,相同数据量重复率越低,后续可用的数据量越多;
Mismatch rate:碱基的错配率;
Average sequencing depth:有效平均深度(不计算duplication),比对到参考基因组的碱基数目除以基因组的大小;目前行业对外承诺的30X(90G)、40X(120G)等深度只是测序量的简单换算,并不是指有效深度。
Coverage at least 1X(4X、10X、20X):覆盖率,指测序深度达到1X、4X、10X、20X以上的全基因组占比。
- 高灵敏度和精准度
高高灵敏度(Sensitivity)和高精准度(Precision)意味着DNBSEQ平台检测发现变异的能力更强,并且结果中为真的突变的概率也高。
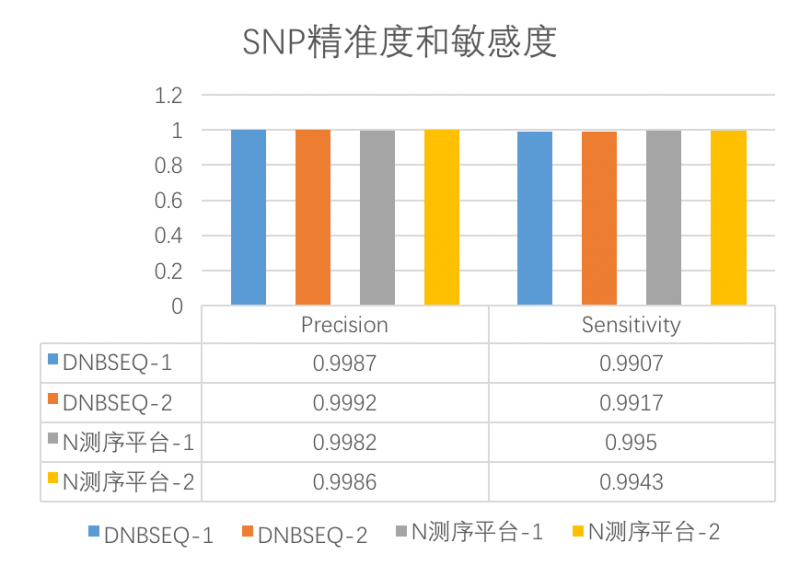
图DNBSEQ与N平台SNP精准度和敏感度对比
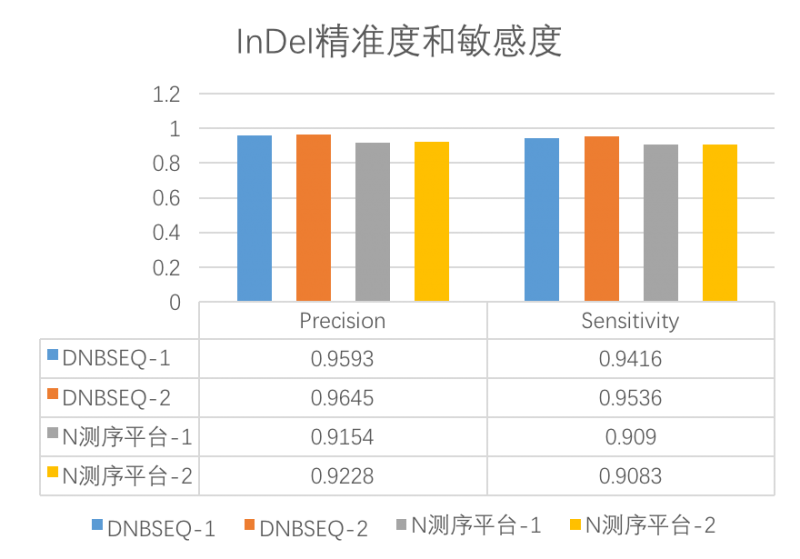
图 DNBSEQ与N平台InDel精准度和敏感度对比
Sensitivity:灵敏度,又叫真阳性率(TPR),计算公式:灵敏度=真阳性/(真阳性+假阴性)。是指实际为阳性的样本中,判断为阳性的比例。例如,真正突变中,被判断为有突变的比例,它反映筛检发现变异的能力,灵敏度越高,假阴性越低;
Precision:精准度,也叫阳性预测值(PPV),计算公式:精准度=真阳性/(真阳性+假阳性),指筛检试验检出的全部阳性变异中,真正“变异”的例数(真阳性)所占的比例,反映筛检变异结果阳性中为真的突变的可能性,精准度越高,假阳性越低。
- 高变异结果一致性
DNBSEQ平台内SNP一致性99%,与N平台间一致性高达98.67%。对于InDel检测一致性,DNBSEQ平台内重复一致性87.94%,远高于N平台内重复一致性79.43%。两平台间的InDel一致性81.98%。
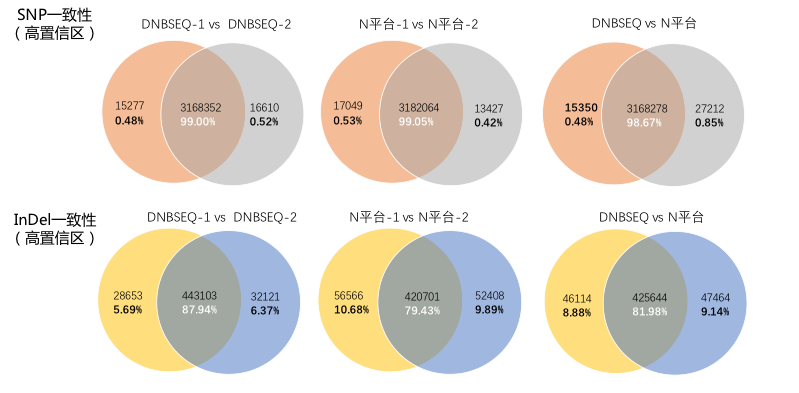
图 平台内与平台间的SNP/InDel一致性对比
2、已交付商业样本数据展示
目前华大基因已成功交付上万例高质量的BGISEQ平台 WGS数据,并得到了海内外业界高度认可。其中包括贝勒医学院(Baylor College of Medicine)、华盛顿大学(University of Washington)、斯坦福大学(Stanford University)、麻省理工(Massachusetts Institute of Technology)等早期参与人类基因组计划(Human Genome Project, HGP)的主要单位,以及牛津大学(University of Oxford)、梅奥诊所(Mayo Clinic.)、康奈尔大学(Cornell University)、费城儿童医院(CHOP)、德国癌症研究中心(German Cancer Research Center)、中南大学湘雅医院、同济医院、清华大学等上百家全球知名科研单位参与平台测试。通过对不同样本类型测试和不同测序平台比较,均获得较高的数据质量结果。在这里我们随机统计了去除样本背景信息后的1,355个样品下机数据,统计具体的质量表现。
- 样本类型适用广泛
DNBSEQ平台 WGS数据来源样本种类多样,其中包含福尔马林固定石蜡包埋( Formalin Fixed and Paraffin Embedde, FFPE)样品、单细胞样品、血液样品、基因组DNA样品、唾液样品、常规冷冻保存的新鲜组织样品等,不同样本类型均有较高的交付成功率,基于BGISEQ平台交付的样本中,常规基因组建库测序成功率高达99%,对于降解样品如FFPE等,建库测序成功率也高达90%以上。
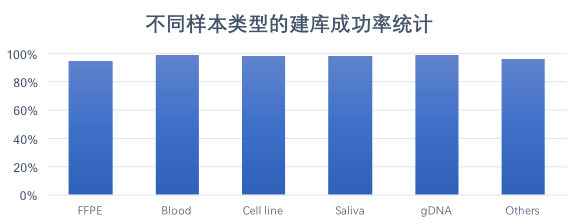
图 DNBSEQ平台 WGS不同类型样品交付成功率
- 数据利用率高
随机统计了1,100条lane的DNBSEQ平台 WGS 下机数据,利用率平均高达99%。
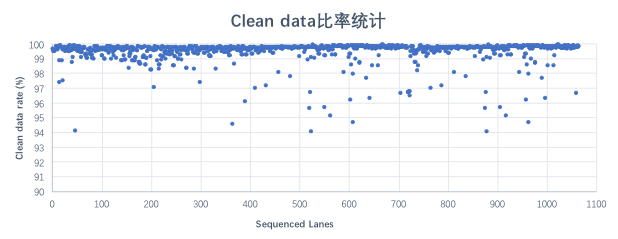
图 Clean data比率
- 测序数据质量优
对随机挑选的1,355条lane DNBSEQ平台 WGS数据质量值进行统计分析,下机Raw data Q20平均值为96.16%,Raw data Q30平均值为87.86%。
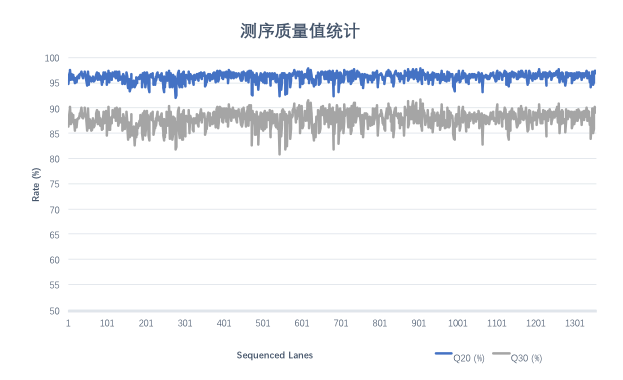
图 碱基质量分布
- GC含量稳定
对该1,355条lane数据的GC含量进行统计分析,平均GC含量为41.69%, GC含量稳定,没有偏向性。
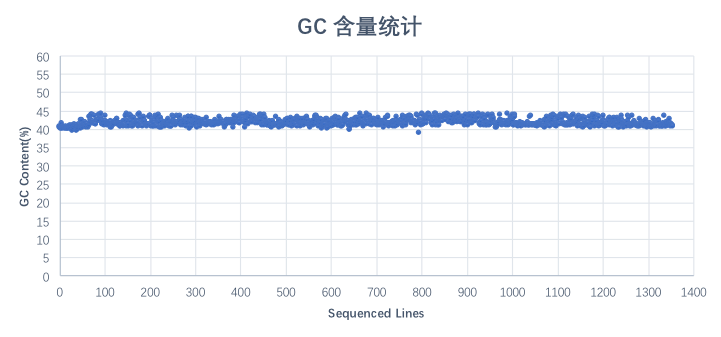
图 GC含量分布
*上述分析结果由华大信息分析流程所得,本结果不代表交付指标,最终解释权归深圳华大基因股份有限公司所有
华大基因作为全球领先的基因组学研究中心及临床解读中心,推出的自主研发的DNBSEQ平台 30X WGS测序在成本和技术上极大的促进了基因组学的快速发展,使基因组学真正的进入了百元基因组时代。DNBSEQ平台见证了人类基因组计划以来一个新时代的开启,将推动以基因测序作为支撑的生命科学、生物产业甚至生命经济蓬勃发展,以其低廉的成本、高质量、高通量的测序平台,真正实现人类基因组计划以来科学家们的梦想和希望!
参考文献:
[1] Ewing B, Hillier L D, Wendl M C, et al. Ewing B, Hillier L, Wendl MC et al.Base-calling of automated sequencer traces using PHRED. I. Accuracy assessment. Genome Res 8:175-185[J]. Genome Research, 1998, 8(3):175-185.
[2] Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities[J]. Genome Research, 1998, 8(3):186-94.
[3] Carrick D M, Mehaffey M G, Sachs M C, et al. Robustness of Next Generation Sequencing on Older Formalin-Fixed Paraffin-Embedded Tissue[J]. Plos One, 2015, 10(7):e0127353.
表1 DNA样本送样建议
|
样本类型
|
总量
|
浓度
|
完整性(胶图)
|
纯度
|
|
Genomic DNA |
≥1μg |
≥12.5ng/μL |
主峰>20Kb |
无蛋白,RNA/盐离子等污染,样本无色透明不粘稠 |
表2 组织样本送样建议
|
组织类型
|
需求量 |
|
新鲜培养细胞 (细胞数) |
≥5×106cell |
|
新鲜动物组织干重
|
≥50mg |
|
全血(哺乳动物)
|
≥1 mL |
|
FFPE |
≥ 10 片,未染色,100 mm2,5 ~ 10μm厚度 |
Q1:DNBSEQ人全基因组重测序的数据格式是否与Illumina平台的一致?
答:是一致的,所以信息分析流程都一样。
Q2:想对我们DNBSEQ产出的数据先分析确定一下格式和数据质量,现在是否有测试过的数据可以提供?
答:DNBSEQ demo数据已经开放,可以访问并下载,链接如下:
表1 上传数据对应ID
|
数据类型 |
Project
Accession ID |
Sample
Accession ID |
Experiment
Accession ID |
|
WGS PCR PE100 |
CNP0000368 |
CNS0050043 |
CNX0043488 |
|
WGS PCR PE150 |
CNP0000368 |
CNS0050043 |
CNX0043489 |
|
WGS PCR-free PE100 |
CNP0000368 |
CNS0050043 |
CNX0043490 |
|
WGS PCR-free PE150 |
CNP0000368 |
CNS0050043 |
CNX0043491 |
Q3:如何实现基因组变异可视化?
答:基因组可视化软件 IGV(Integrative Genomics Viewer)是高性能的基因组数据可视化工具,能够帮助使用者同时合并分析不同类型的基因组数据,并能灵活放大基因组上的某个特定区域。IGV 软件免费下载地址: http://www.broadinstitute.org/igv. IGV 可查看 SAM / BAM 比对文件和 VCF 变异检测文件,下图显示的是 IGV 可视化窗口。
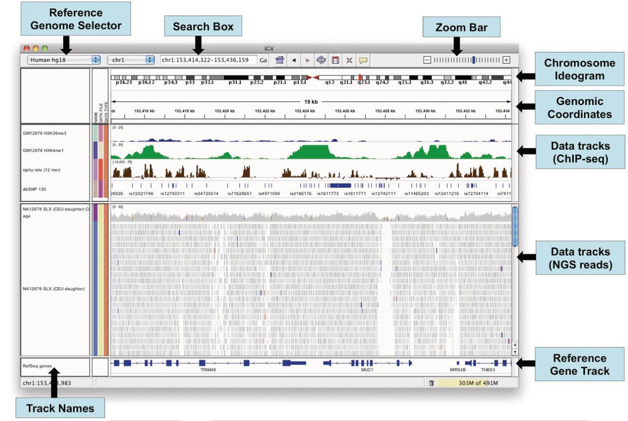
图1 IGV可视化窗口示意图
Q4:如何寻找候选变异?
答:寻找候选变异位点时,可利用变异注释结果,关注非同义突变、剪接突变、移码突变。1)去除千人基因组数据库中 MAF >=1% 的变异2)去除 NHLBI-ESP6500 European American 群体数据库中 MAF >=1% 的 变异 3)去除 NHLBI-ESP6500 African American 群数据库中 MAF >=1%的变异4) 推 测 变 异 的 致 病 性 。 利 用 SIFT/PolyPhen2/Mutation assessor/Condel/FATHMM 进行打分,预测某个变异和氨基酸置换是否影响蛋白 功 能 。 如 果 score<=0.05 或 PolyPhen2>=0.909 或 MA score>=1.9 或 Condel = deleterious 或 FATHMM=deleterious,就推测该变异可能是有害变异。
Q5:SNP 筛选所使用的数据库有哪些,怎么筛选?
答:数据库: dbSNP 、 HapMap3、 1000 Genomes 一般情况下,我们都采用以下过滤标准: 1、质量值不低于 20; 2、覆盖深度不低于 4; 3、两个相邻 snp 之间的距离不小于 5,如果样本深度很高(>50X),可以提高过滤条件。
Q6:一般用什么方法来验证 call SNP 准确率?
答:华大炎黄计划是用 Sanger 测序的方法和芯片分型两种方法来验证 SNP 的准确性的, 因为 Sanger 测序被认为是测序中的“金标准”。
Q7:唾液采集的方法?
答:使用DNA Genotek公司的 Oragene•DISCOVER (OGR-500) (For Research) 或 Oragene•Dx (OGD-500) (For Diagnostics) collection kit.保存量及操作方法详见产品说明书,按照说明书操作保存运输样品。
Q8:突变位点为有效位点时使用的 depth 阈值是多少?
答:GATK在call变异时SNP和InDel均要求depth大于等于4 。
Q9:数据中的 Duplicates 指什么?如何定义?有何影响?
答:一般情况下,测序得到了两对或两对以上的pair end reads同时比对到参考序列上相同的起始和结束位置,我们定义这种序列为duplicates。
在数据分析过程中,为了确保变异分析的准确性,避免计算存储资源的浪费,一般会通过生信的方法去掉Duplicate reads后再进行下游信息分析。
但这么做,至少会带来以下2方面的问题:
1、 数据量浪费
越高的duplicates比例,为此而浪费的数据量就越大。按照illumina平台为例,普遍的duplicates比例大约在10%左右。也就是花了100G data的钱,有用的只有90G左右。
2、 对于RNA-Seq,无法去除
对于RNA来说,因为难以区分是PCR duplicates还是RNA高表达形成的相同的模板,则无法去除duplicates。从而影响转录组表达量的准确性,尤其是小和中等表达量的转录本的准确性。