- 首页 > 动植物de novo测序
动植物de novo测序
动植物de novo测序即动植物从头测序,指不需要任何参考序列信息即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动植物的全基因组序列,带动这个物种下游一系列研究的开展,从而推进该物种的研究。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台,为后续的基因挖掘、功能验证提供DNA序列信息。
产品优势
测序通量高:30+DNBSEQ、1台PacBio Sequel 、1台PacBio Sequel II、2台 Nanopore PromethION测序仪保证超高测序通量。
测序质量好:每种测序平台均执行严格质控,保证测序质量。
平台多样化:PacBio/Nanopore/DNBSEQ/Hi-C/stLFR多技术平台,为基因组组装提供整体解决方案。
应用范围广:基因组图谱的完成为后续基因挖掘、物种起源及进化等研究提供大量数据支撑。
项目经验足:分析人员从业时间久、资历深、经验丰富,精通基因组产品相关的各种分析,为项目的顺利交付保驾护航。
结果产出高:华大基因已经成功完成1,000多个物种的全基因组从头测序,合作发表动植物de novo文章310篇,封面文章29篇。
产品应用
- 获得物种的参考序列
- 研究物种起源与进化历史
- 挖掘功能基因
- 搭建物种数据库
研究内容
基因组Survey:
通过多个Kmer估计基因组大小和基因组杂合率,重复水平(软件Jellyfish+genomeScope)
基因组组装(PacBio HiFi):
1. 组装
2. BUSCO评价
基因组组装(PacBio CLR/Nanopore+NGS):
1. repeat注释
2. 基因结构注释(建议提供同源物种 5-6 个以及转录组数据)
3. 基因功能注释
4. 转录因子预测(植物)
进化分析:
提供已发表物种和近缘物种(选择 10 个物种以内)
1. 因家族鉴定(OrthoMCL/OrthoFinder)
2. 物种系统发育树构建(phyml, Bayers, RAxML, iqTree)
3. 物种分歧时间估算(PAML)
4. 基因组共线性分析(植物,物种自身 MCScanX;物种间JCVI)
5. 全基因组复制分析(植物,Ks,4DTv)
6. 基因家族扩张收缩分析(CAFÉ)(结果有无取决于分歧时间差异大小,分歧时间差异较大无结果)
Hi-C辅助组装:
1. 文库评估
2. Hi-C分析
3. 手工矫正,获得染色体
4. 近缘物种比较,染色体定名
定制化信息分析:
可结合客户的需求,协商确定定制化信息分析内容
案例一:多平台构建鸭嘴兽和短鼻针鼹的高质量基因组
生活在澳洲大陆的鸭嘴兽和针鼹,组成了哺乳动物原兽亚纲的单孔目。相比其他哺乳动物,单孔目有很多独特之处,如单孔目动物排尿、排便、生殖都共用一个孔,这也是单孔目得名的由来。不仅如此,单孔目动物的染色体结构也异乎寻常:包括人类在内的其他哺乳动物一般只有一对性染色体,雌性XX和雄性XY,而单孔目动物却有5对10条性染色体。单孔目动物凭借其非凡的生物学特性和独特的演化地位,在哺乳动物起源与演化研究中备受科学家青睐。
1. 构建染色体级别的高质量基因组
基于PacBio、Bionano、Hi-C等最新测序方法,华大与合作伙伴为鸭嘴兽和针鼹这两个物种构建了染色体级别的高质量基因组,为后续研究内容的开展提供了坚实的数据支撑。
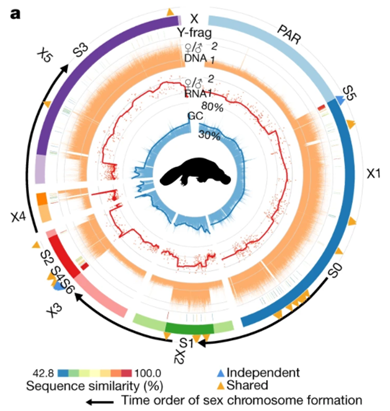
图 鸭嘴兽性染色体组装及其特性
2. 构建出现生哺乳动物的祖先染色体
对于现生哺乳动物的共同祖先,能通过化石证据还原它们的一部分外观特征,但其染色体数目尚不清楚。基于本项研究产生的两个高质量单孔目基因组,研究团队首次构建出现生哺乳动物最近共同祖先染色体(2n=60),绘制了现生哺乳动物早期祖先的演化图谱。现生哺乳动物的共同祖先生活在距今约1.8亿年前,染色体数为60条;兽亚纲哺乳动物共同祖先出现在距今约1.6亿年前,染色体数为36条;有袋类哺乳动物祖先出现在距今约8千万年前,染色体数为14条。
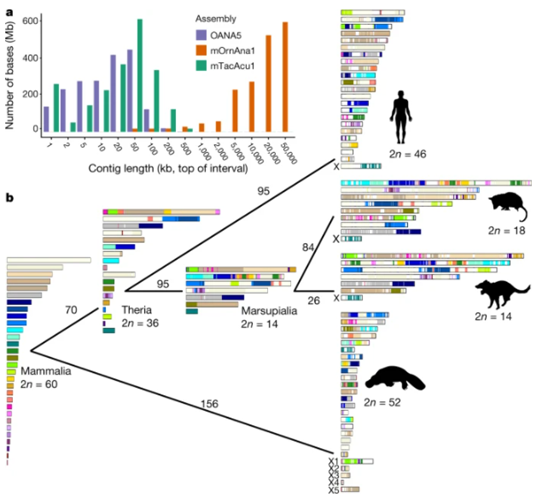
图 哺乳动物核型进化轨迹
3. 单孔目的性染色体形成机制
研究团队发现,单孔目物种的5条X染色体与其他绝大多数哺乳动物的X染色体序列并不相似,但有一部分序列跟鸟类的性染色体同源。进一步与其它物种基因组进行比较分析,研究人员发现,此前针对单孔目性染色体形成机制的主流假设——5对性染色体由两个古老的单孔目类群杂交产生——并不成立,提出单孔目中的多对性染色体更可能是通过多对古老的常染色体相互之间发生了非同源片段的交换,即转座异位事件,在多次交换之后,形成了如今的5对性染色体。并推测在单孔目性染色体演化的初期,5对性染色体可能在减数分裂时形成一个首尾相接的罕见环状结构,但这个结构随着Y染色体的一步步退化最终断开,形成了我们现在观测到的链状结构。
4. 单孔目动物独特的生物学特性相关基因
研究团队分析发现,单孔目动物与人、考拉等哺乳动物不同,保留了一部分与鸟类和爬行类相似的基因,这些基因参与卵的形成;相比于鸟类和爬行类,单孔目物种已经拥有一些参与乳汁分泌过程的基因,如合成乳汁中的主要成分酪蛋白的基因;成年的鸭嘴兽和针鼹没有牙齿,胃也基本退化,研究发现在鸭嘴兽和针鼹基因组当中,许多与牙齿和胃发育相关的基因已丢失,比如与牙齿形成、生长以及牙釉基质矿质化过程相关的odontogenic ameloblast-associated基因,以及引导ATP酶将氢离子泵入胃中、刺激胃酸分泌的gastrin胃泌素基因,均已丢失;针鼹的嗅觉受体基因明显多于鸭嘴兽和其他哺乳动物;而在鸭嘴兽基因组当中,与夜行性相关的犁鼻器受体基因数量则明显更多。
本研究不仅突破了性染色体组装的技术难题,构建得到了优质的单孔目参考基因组,还通过精妙的数据分析,追溯了距今1.8亿年前现生哺乳动物共同祖先的染色体图谱,帮助学界更好地理解物种演化过程的分子机制,此外也找到了特定的基因,解释了单孔目动物独特生物学特性的产生机制。
参考文献
Zhou Y, Shearwin-Whyatt L, Li J, et al. Platypus and echidna genomes reveal mammalian biology and evolution[J]. Nature, 2021: 1-7.
1. 基因家族鉴定
通过同源基因的鉴定及基因家族的聚类分析,得到保守的单拷贝基因家族和多拷贝基因家族,以及物种特有的基因和家族,它们可能和物种的特异性有关,可以为物种特性的研究提供基础。通过Orthofinder软件对蛋白基因集进行聚类得到基因家族信息。
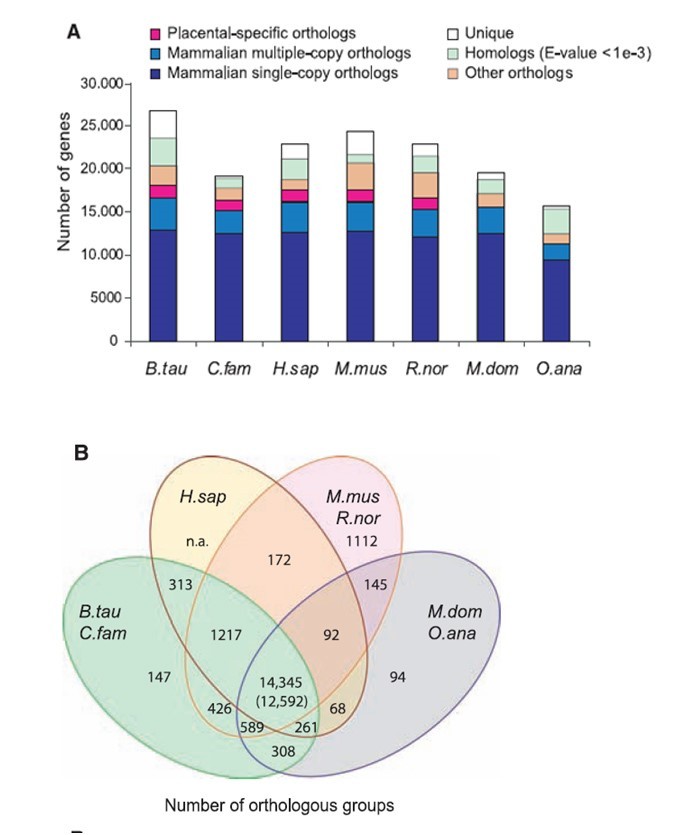
图1:A图表示不同物种间直系同源基因的种类及数量; B图表示不同物种间直系同源基因的种类及数量韦恩图;
2. 系统发育分析
利用单拷贝基因家族构建物种发育树。根据基因家族聚类的结果,使用单拷贝直系同源基因利用MUSCLE 、Gblocks 0.91b、RaxML软件进行多序列比对,提取保守区域,构建进化树,并使用FigTree进行定根。
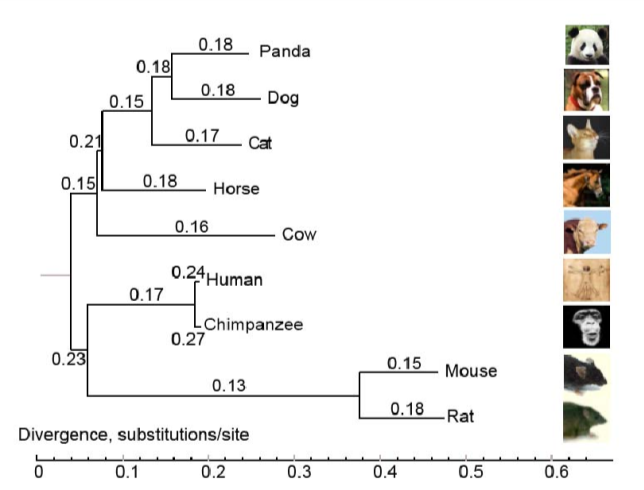
图2:系统发育树
3. 物种分化时间估算
通过每个单拷贝基因家族中的简并位点、系统发育中的定根树及已知物种的分化时间,使用PAML软件估算分子钟和物种间的分化时间。
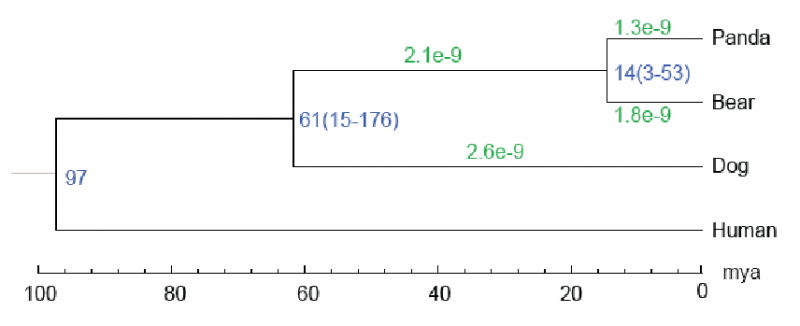
图3 物种分化时间。每个分枝长度代表中性进化速率,树形结构节点处数字表示支持率
4. 基因家族扩张与收缩分析
通过基因家族的信息、计算得到的系统发育树和物种分化时间来进行基因家族的扩张与收缩分析。
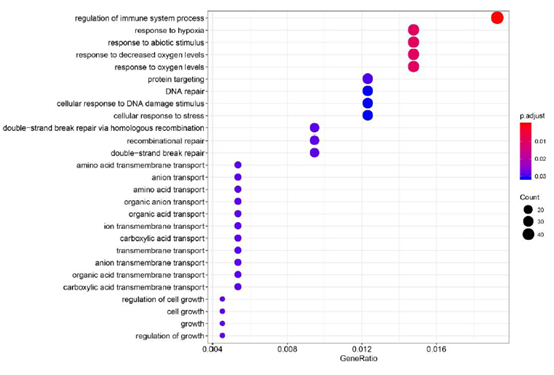
图4 扩张与收缩的基因家族GO功能富集
5. 基因组共线性分析
共线性片段指同一个物种内部或者两个物种之间,由于复制(基因组复制、染色体复制或者大片段复制)或者物种分化而产生的大片段的同源性现象。在共线性片段中的基因在物种进化过程中保持了高度的保守性。现在常采用 MCScan、MCScanX或JCVI软件进行分析。

图5 自身共线性分析
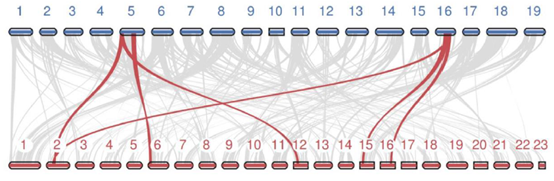
图6 物种间共线性分析
6、全基因组复制分析(ks)
Ks分析物种在进化史中是否发生全基因组复制事件、以及通过它与其它植物分化时间的比较区分发生全基因组复制相对时间的早晚。将筛选到的共线性基因及其比对结果利用PAML软件对每个基因对进行Ks计算,推断物种分化时间节点或者全基因组复制时间。
各个平台DNA送样要求
|
平台 |
文库类型 |
样本量(基因组DNA) |
浓度 |
|
Nanopore PromethION |
20-40Kb library |
≥9μg |
90 ng/μl |
|
Ultra-long library(>40k) |
≥10μg |
150 ng/μl |
|
|
PacBio Sequel II |
15-20Kb HIFI library |
≥15μg |
80 ng/μl |
|
20-40kb CLR library |
≥7μg |
70 |
|
|
DNBSEQ |
350bp library |
≥1μg |
12.5 ng/μl |
|
StLFR library |
≥500ng |
1 ng/μl |
注:大片段文库不建议客户送DNA样本,建议直接送组织,详细组织送样建议请联系当地销售。
Q1:怎么查询基因组的大小?
答:查询植物基因组大小的网站:http://data.kew.org/cvalues/CvalServlet?querytype=2;
查询动物基因组大小的网站:http://www.genomesize.com/search.php。
换算关系:1pg=978Mb。
Q2:基因组从头测序的组装结果好坏如何判断?
答:一般用contig N50和scaffold N50 来衡量基因组组装结果的好坏。N50是指把组装出的contigs或scaffolds从大到小排列,当其累计长度刚刚超过全部组装序列总长度50%时,最后一个contig或scaffold的大小即为N50的大小,N50对评价组装序列的连续性、完整性有重要意义;N70和N90的计算方法与N50类似,只是百分数变为70%或90%。
Q3:PacBio测序的优势是什么?
答:优势是测序读长长,平均读长在15K以上,且无GC偏向性;对基因组的组装、大的结构变异检测、转录组全长测序结果均有极大提升。
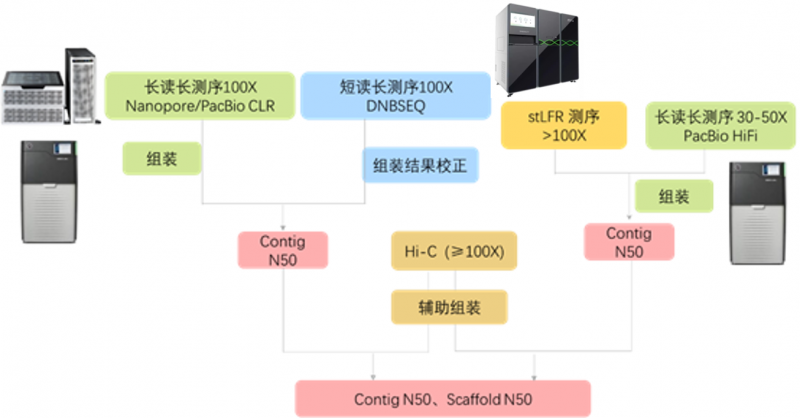
Q4:进行基因组组装有哪些测序策略推荐?
答:以下是我们推荐的基因组组装策略,我们的技术团队会根据物种特性推荐合理的方案。