- 首页 > 真核转录组测序
真核转录组测序
转录组测序,对某一物种或特定细胞在某一功能状态下产生的mRNA进行高通量测序,既可以提供定量分析,检测基因表达水平差异,又可以提供结构分析,能发现稀有转录本,精确地识别可变剪切位点、基因融合等,而且不依赖于参考基因组。
技术优势
任意物种的全转录组分析:无需预先设计特异性探针,因此无需了解物种基因或基因组信息,能够直接对任何物种进行全面的转录组分析;
覆盖度高:数字化信号,直接测定几乎所有转录本片段的序列;
检测阈值宽:跨越6个数量级的宽检测阈值,从几个到数十万个拷贝精确计数;
分辨率高:可以检测基因家族中相似基因及可变剪接造成的单碱基差异;
检测范围广:从几个到数十万个拷贝精确计数,可同时鉴定及定量正常和稀有的转录本。
产品应用
医学上的应用方向——
农学上的应用方向 ——
研究内容
一、无参考序列物种
标准信息分析:
1. 测序数据过滤;
2. 转录组de novo组装;
3. Unigene七大功能数据库注释;
4. Unigene的CDS预测;
5. Unigene的TF编码能力预测;
6. Unigene的SSR检测;
7. Unigene表达量计算(基因表达水平、PCA分析、基因表达水平箱线图和密度图、样品间的表达量韦恩图);
8. 时间序列分析;
9. 差异表达基因检测;
10. 差异表达基因聚类分析;
11. 差异表达基因GO功能分析;
12. 差异表达基因Pathway功能分析;
13. 差异基因蛋白互作分析;
14. 真菌致病基因预测 (真菌样本);
15. 植物抗病基因预测(植物样本)。
定制化信息分析:
1. 可结合客户的需求,协商确定定制化信息分析内容。
二、有参考序列物种
标准信息分析:
1. 基本数据统计① 去除接头序列、低质量序列得到reads信息,② 样品相关性,③ 表达量分布,④ RNA分类;
2. 参考基因组比对;
3. mRNA鉴定;
4. mRNA定量分析;
5. mRNA差异表达分析(样本间、组间);
6. mRNA表达/差异基因聚类;
7. mRNA差异基因GO分类、富集;
8. mRNA差异基因KEGG分类、富集;
9. mRNA结构分析:可变剪切分析;
10. mRNA结构分析:融合基因分析(仅限人)。
Dr.Tom信息分析:
一)数据库注释
1. 转录因子注释(AnimalTFDB/PlantTFDB);
2. GSEA分析;
3. Rfam、Pfam、Reactome、COG、EggNOG和InterPro数据库注释;
二)互作网络分析
1. 靶基因分析① miRNA-mRNA靶向关系分析,② lncRNA-mRNA靶向关系分析;
2. ceRNA互作网络分析;
3. 蛋白互作网络分析;
4. 共表达互作网络分析;
三)特色分析
1. 自定义标签和自有数据上传;
2. 外部数据库信息(TCGA、ARCHS4);
3. 关键驱动基因网络图分析;
4. 时间序列分析。
(*以上分析内容为部分物种可做。)
定制化信息分析 :
1. 可结合客户的需求,协商确定定制化信息分析内容
医学案例:转录组研究抑癌基因失活将导致胃肠道间质瘤恶性进展
文章亮点:
首次解析了胃肠道间质瘤恶性进展的新机制,鉴定出抑癌基因DEPDC5 。
文章概述:
DEPDC5抑癌基因失活将导致胃肠道间质瘤恶性进展。
研究团队在国际上首次解析了胃肠道间质瘤恶性进展的新机制,鉴定出位于22号染色体的主要抑癌基因DEPDC5,解析了DEPDC5 抑癌基因的失活是间质瘤恶性进展的重要分子机制,提供了区分间质瘤恶性程度的分子标记物,为间质瘤的精准治疗提供了实验依据。
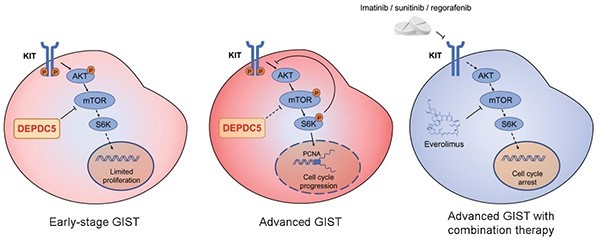
图1 DEPDC5 抑癌基因的失活促进胃肠道间质瘤恶性进展
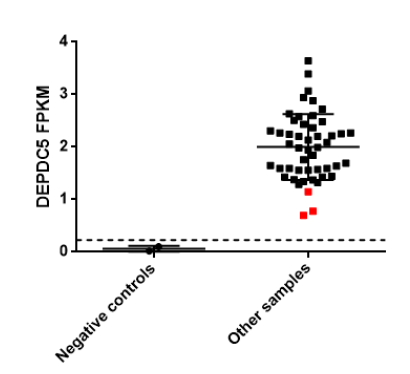
图2 转录组测序检测胃肠道间质瘤中DEPDC5表达
参考文献:
Pang Y, Xie F, Cao H, et al. Mutational inactivation of mTORC1 repressor gene DEPDC5 in human gastrointestinal stromal tumors[J]. PNAS, 2019, 116(45): 22746-22753.
农学案例:转录组测序构建大鲵的参考基因集
研究目的:构建大鲵完整的参考基因集。
研究材料:收集成年中国大鲵的20多个组织样本(腹部皮肤、背部皮肤、侧部皮肤、肺、心脏、肾、胰腺、小肠、脾脏、胃、脑、脊髓、软骨、眼睛、指尖、长骨、上颌骨、头盖骨、肌肉、 卵巢、脂肪、尾部脂肪、血液)。
研究方法:转录组测序,每个样本约为6.5Gb。
研究结果:
1. 为了获得一个完整的参考基因集,将所有样本的clean reads混合在一起组装得到93,366条非冗余转录本,平均长度1,326bp。
2. 将组装得到的序列注释到NR、Swiss-Prot,KEGG、COG、GO数据库进行功能注释,最后有41,874条序列被注释到。
3. 采用BlastX,ESTscan、Transdecoder 软件分别预测CDS序列,然后再进行整合(至少2种方法支持),并且需要CPC≥1,最后鉴定了26,135个蛋白。
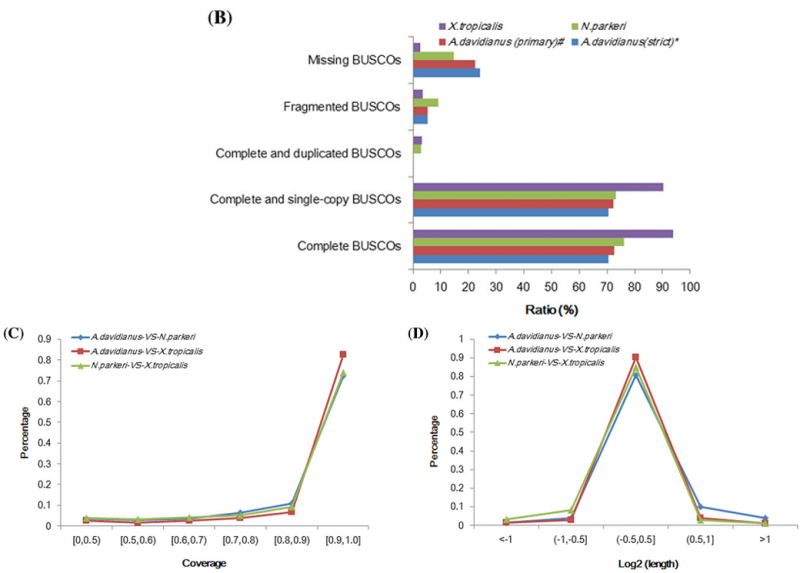
图2 转录组组装评估
B: BUSCO评估基因集、C和D:和热带爪蟾、高山倭蛙比较CDS长度
参考文献:
Geng X, Li W, Shang H, Gou Q, et al. A reference gene set construction using RNA-seq of multiple tissues of Chinese Giant Salamander, Andrias davidianus. Gigascience. 2017 Feb 15. doi: 10.1093/gigascience/gix006.
部分内容展示:
图1 差异聚类热图
图2 功能富集或分类图

图3 蛋白互作网络
表1 真核转录组测序核酸样品判定标准
|
真核转录组(默认链特异性,非链特异和链特异性送样建议相同) |
||||||
|
样本类型 |
总量 |
浓度 |
RIN |
28S/18S (23S/16S) or DV200 |
基线和 5S |
纯度 |
|
Total RNA(真菌) |
≥1μg |
20-1000 ng/μL |
RIN≥6.5 |
28S/18S≥1.0 |
基线平整, 5S 峰正常 |
无 DNA,蛋白/盐 离子等污染,样本 无色透明不粘稠
|
|
Total RNA (植物) |
≥400ng |
10-1000 ng/μL |
RIN≥6 |
28S/18S≥1.0 |
||
|
Total RNA(动物) |
≥400ng |
10-1000 ng/μL |
RIN≥7.0 |
28S/18S≥1.0 |
||
|
Total RNA (非全血人鼠)
|
≥200ng |
10-1000 ng/μL |
RIN≥7.0 |
28S/18S≥1.0 |
||
|
Total RNA(昆虫) |
≥400ng |
10-1000 ng/μL |
N/A |
|||
表2 真核转录组测序组织样品判定标准
|
组织类型 |
真核转录组文库 |
|
新鲜培养细胞 (细胞数) |
≥2×105cell
|
|
新鲜动物组织干重 |
≥30mg
|
|
新鲜植物组织干重 |
≥100mg
|
|
全血 |
≥1
mL 全血收集的淋巴细胞或 ≥1
mL Paxgene Blood RNA tube / RNAprotect ®
Animal Blood Tubes收集的全血 |
|
菌体(细胞数或干重) |
≥2×105cell
或 ≥30mg |
|
FFPE |
≥5片,未染色,100mm2,5~10μm 厚度 |
Q1:BGISEQ-500滚环扩增技术的特点是什么?
答:滚环扩增技术RCA的模板始终是同段序列,扩增错误不会累积,与PCR指数扩增相比于保真优势。
Q2:BGISEQ-500平台的测序原理是什么?
答:BGISEQ-500采用优化的联合探针锚定聚合技术(cPAS)和改进的DNA纳米球(DNB)核心测序技术,是行业领先的高通量测序平台之一。具体而言,首先DNA分子锚和荧光探针在纳米球上进行聚合,随后高分辨率成像系统对光信号进行采集,光信号经过数字化处理后即可获得待测序列。其中,DNB通过线性扩增增强信号,降低单拷贝的错误率。而且,DNB大小与芯片上活性位点的大小相匹配,每个位点结合一个DNA纳米球,在保证测序精度的情况下提高了测序芯片的利用效率。
Q3:BGISEQ-500 下机数据格式是什么?
答:通用下机数据格式:(FASTQ)让数据兼容性更好。
Q4:BGISEQ-500平台真核转录组推荐数据量?
答:推荐6G或者10G数据量。
Q5: 可否提供BGISEQ-500转录组标准品测试数据供客户测评?
答:可以,我们 1个UHRR标准品,构建了3个文库,数据都已经上传到EBI。请点击下面链接下载。http://www.ebi.ac.uk/ena/data/view/PRJEB19428。
Q6: 可否提供BGISEQ-500真核转录组实验流程英文版?
答:The first step in the workflow involves purifying the poly-A containing mRNA molecules using poly-T oligo-attached magnetic beads. Following purification, the mRNA is fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments are copied into first strand cDNA using reverse transcriptase and random primers. This is followed by second strand cDNA synthesis using DNA Polymerase I and RNase H. These cDNA fragments then have the addition of a single 'A' base and subsequent ligation of the adapter. The products are then purified and enriched with PCR amplification.
We then quantified the PCR yield by Qubit and pooled samples together to make a single strand DNA circle (ssDNA circle), which gave the final library.
DNA nanoballs (DNBs) were generated with the ssDNA circle by rolling circle replication (RCR) to enlarge the fluorescent signals at the sequencing process. The DNBs were loaded into the patterned nanoarrays and pair-end read of 100 bp(or 150 bp) were read through on the BGISEQ-500 platform for the following data analysis study. For this step, the BGISEQ-500 platform combines the DNA nanoball-based nanoarrays and stepwise sequencing using Combinational Probe-Anchor Synthesis Sequencing Method.