- 首页 > 代谢组+宏基因组关联分析
代谢组+宏基因组关联分析
微生物组的研究已经逐渐进入到多组学的时代,越来越多的科学家将代谢组与微生物组关联起来,同时从微生物与代谢物两个角度分析生物学问题,通过解析微生物-代谢物的关联性,从多维的角度揭示生命活动。
华大基因针对宏基因组及代谢组的特征数据差异物种及差异代谢物进行整合分析——对于差异微生物及差异代谢物分别进行定量关联分析及功能关联分析,对于提供连续表型的数据,还进行差异物种,差异代谢物和表型之间的关联分析,以探究微生物、代谢物和表型的内在关系。
技术优势
1、关联分析研究经验丰富,在Nature 等顶级期刊上自主发表多篇文献。
2、充分调研微生物组与代谢组关联分析文献,自主开发软件分析流程。
产品应用
1、 肠道微生物与疾病研究
2、 脑-肠-轴研究
3、 益生菌等微生物制剂开发
4、 根际微生物研究
5、 水体、土壤等环境研究
6、 茶叶、腐乳等发酵食品发酵工艺探索
技术路线
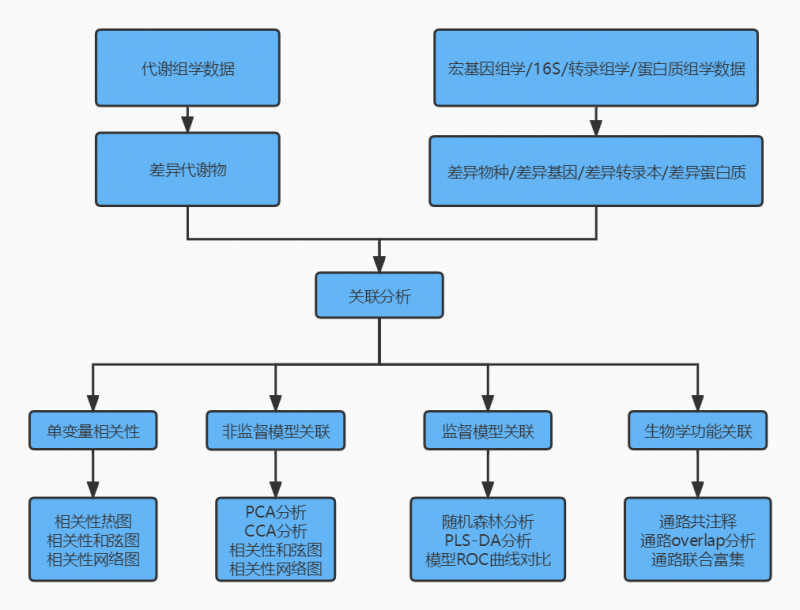
注:差异物种无生物学功能关联
项目周期
12个自然日
案例一:胆汁酸检测联合宏基因组探究益生菌及小檗碱对初发2型糖尿病的治疗作用[1]
研究背景:
在过去的十多年中,研究发现肠道微生物是一个有前景的2型糖尿病(T2D)治疗靶点,小檗碱(BBR,一种抑菌素)也被发现可以治疗包括T2D在内的一些代谢紊乱的疾病。研究拟通过宏基因组、代谢组等多组学技术探究BBR联合使用益生菌对初发T2D患者的治疗作用。
技术路线:
将409个初发T2D志愿者分为安慰剂组(Plac)、益生菌组(Prob)、小檗碱组(BBR)及益生菌联合小檗碱组(Prob+BBR)进行了为期3个月的临床试验,结合临床医学数据、宏基因组数据及代谢组数据对联合使用益生菌与小檗碱治疗T2D的方案进行了评估。
主要结论:
临床生化指标评估显示包含糖化血红蛋白HbA1c在内的多项T2D相关的生化指标在BBR介入后发生明显改善。宏基因组结果显示,与Plac组相比,BBR对T2D患者的肠道微生物有显著调节作用,且这种调节作用与益生菌的使用无关。血浆胆汁酸检测则表明BBR能通过抑制微生物胆汁酸的转化降低肠道法尼醇X受体(FXR)的活性,促进其抗糖尿病作用。最后体外实验证明BBR能通过抑制R.bromii的生长削弱次级胆汁酸的代谢,从而起到调节血糖的作用。
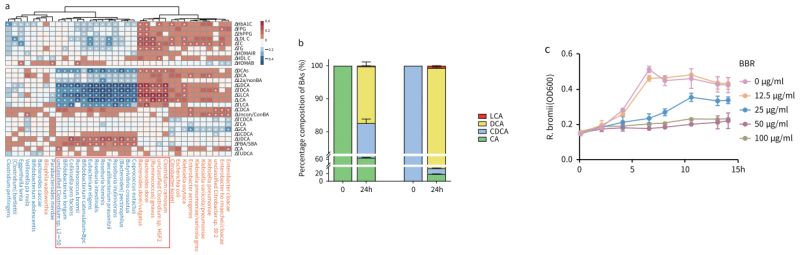
图1 BBR通过抑制R.bromii消弱DCA的转化
参考文献
[1]Gut microbiome-related effects of berberine and proloiotics on type 2 diabetes(the PREMOTE study) Nature communications 2020
在关联分析中,综合运用了单变量相关性、非监督模型关联、监督模型关联、生物学功能关联。以下为部分分析结果图片展示:
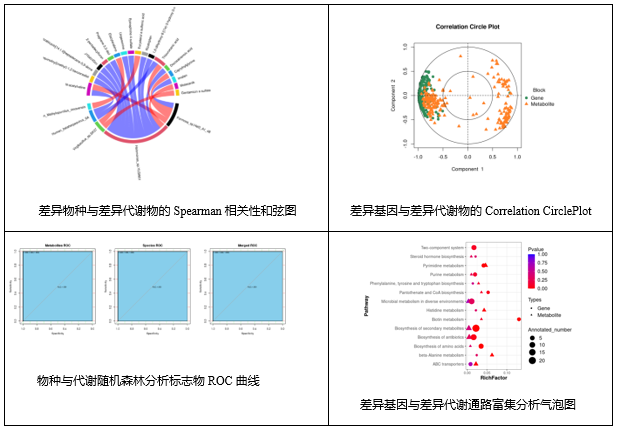
1、 适用数据类型
要求用于关联分析的两组学的样本数据一一对应(即用于关联分析的两组学的样本来自于相同的生物学重复个体),代谢组数据可以为非靶向代谢组、脂质组学、靶向代谢组学中的任意一种。
2、 重复次数要求
1)当样本为2组时,每组样本数不少于8个;当样本组不少于3组时,每组样本不少于6个;每组样本大于10个时分析效果较好;
2)用于关联分析的临床样本推荐每组样本数不少于30个;
3)重复数少于建议数时分析效果差;每组低于3个样本时无法分析。
Q1:为什么用于关联分析的两组学样本要求一一对应?
A1: 分析中包含Spearman相关性分析,其相关系数表明X(独立变量)和Y(依赖变量)的相关方向,在进行计算两组学的相关性时要求每一个组学数据要有能够对应的另一个组学数据,否则无法进行相关性分析。
Q2:宏基因组+代谢关联分析对用于采集数据的样本有限制吗?
A2:没有限制。此关联分析针对宏基因组及代谢组数据,满足条件的数据即可进行相应的关联分析,根据解读关联分析的结果进行数据挖掘,因此要求在前期设计实验时就要考虑到后面用于关联分析的两组学数据的样本来源在生物学上的相关性(一般建议用于关联分析的两组学数据来源于相同的生物学重复的个体),单纯的数据分析本身只讨论统计学的相关性。
Q3:对于关联分析的两组学数据的重复数的要求有什么依据吗?
A3:建议数是我们的项目经验值,当重复数低于建议数时,数据分析的表现结果较差;且每组低于3个样本时无法进行关联分析。