- 首页 > 16S/18S/ITS扩增子测序
16S/18S/ITS扩增子测序
产品介绍
16S rDNA是细菌分类学研究中最常用的“分子钟”,其序列包含9个可变区(Variable region)和10个保守区(constant region)。可变区因细菌而异,且变异程度与细菌的系统发育密切相关。通过检测16S rDNA的序列变异和丰度,可以了解环境样品中群落多样性信息。基于16S rDNA的分析在微生物分类鉴定、微生态研究等方面起到重要作用。
18S rDNA或ITS(Internal Transcribed Spacer)被广泛应用在真菌分类鉴定中。18S rDNA在系统发育研究中较适用于种级以上阶元的分类;ITS属于中度保守区域,利用它可研究种及种以下的分类阶元。
研究内容
16S/18S/ITS扩增子测序即通过提取环境样品的DNA,选择合适的通用引物扩增16S/18S/ITS的某一或某几个区,使用Illumina测序将目的区域正反向读通,通过检测目的区域的序列变异和丰度,对环境样本物种分类及,丰度,种群结构,系统进化,群落比较等方面信息进行分析的研究方法。
产品优势
策略多样:不同来源样本采用不同提取方法和建库测序策略,满足多种环境研究需求。
平台多样:DNBSEQ/PacBio等多种平台可供选择,可满足16S/18S/ITS不同高变区域或全长测序的需求。
经验丰富:已测序样品类型涉及粪便、土壤、水体、唾液、牙菌斑、体腔、胃液、白带、空气、血液、皮屑等。
分析自动化:自动化分析平台,可多次提交不同的信息分析方案,客户可以主导信息分析过程。
售前、售后服务:提供结题报告解读视频及其他个性化售前售后服务。
样本需求量低:华大基因16S产品推荐DNA样本量50ng以上;对于样本获取困难的样本,只要样本量高于0ng也有可能建库成功。
数据交付指标高:华大基因对16S产品承诺交付clean tags(用于后续OTU/物种分析的有效数据量),承诺100%满足合同数据量。常见的16S数据交付指标有raw reads、clean reads、raw tags,clean tags等,受数据质量和目标区域长度的影响,一般的数据利用率(clean reads/raw reads)和拼接率(clean tags/clean reads)会在50%~100%之间波动。
应用范围
医学领域:人体微生物与人体健康/疾病的关系,人体微生物对疾病干预过程的影响;
动物领域:肠道、瘤胃(如产甲烷菌类群)与动物健康/营养消化研究等;
农业领域:根际微生物与植物互作、农业耕作/施肥处理与土壤微生物群落等;
环境领域:雾霾处理、污水治理、石油降解、酸性矿水处理及海洋环境等;
特殊极端环境:极端环境条件下的微生物类群研究,如冰川、火山等。
技术流程
取质量合格的基因组DNA样品30ng及对应的融合引物配置PCR反应体系,设置对应的PCR反应参数进行PCR扩增,对PCR扩增产物进行纯化,完成建库。使用Agilent 2100 Bioanalyzer对文库的片段范围及浓度进行检测。检测合格的文库在华大自主的DNBSEQ平台上进行测序。下机数据经过数据过滤,滤除低质量的reads,剩余高质量的Clean data方可用于后期分析;通过reads之间的Overlap关系将reads拼接成Tags;在给定的相似度下将Tags聚成OTU,然后通过OTU与数据库比对,对OTU进行物种注释;基于OTU和物种注释结果进行样品物种复杂度分析以及组间物种差异分析。
建库流程
16S/18S/ITS扩增子建库推荐融合引物建库方法,即提前合成融合了目标序列引物和上机接头、index等序列的引物,通过一步PCR扩增直接完成建库。主要步骤如下:
样品检测:使用Qubit对样品浓度进行精确定量,检测合格的样品进行文库构建。
PCR体系构建:取30ng DNA样品及融合引物配置PCR反应体系。
PCR扩增:设置PCR反应参数进行PCR扩增。
产物纯化:使用磁珠进行纯化,文库构建完成。
文库质量检测:文库检测使用Agilent 2100 Bioanalyzer检测文库的片段范围及浓度。
上机测序:文库检测合格,上机测序
测序策略
|
|
扩增区域 |
引物名称 |
引物对 |
|
细菌 |
V4 |
515F |
GTGCCAGCMGCCGCGGTAA |
|
806R |
GGACTACHVGGGTWTCTAAT |
||
|
V1-V3 |
8F |
AGAGTTTGATYMTGGCTCAG |
|
|
518R |
ATTACCGCGGCTGCTGG |
||
|
V3-V4 |
338F |
ACTCCTACGGGAGGCAGCAG |
|
|
806R |
GGACTACHVGGGTWTCTAAT |
||
|
V4-V5 |
515F |
GTGCCAGCMGCCGCGG |
|
|
909R |
CCGTCAATTCMTTTRAGT |
||
|
真菌 |
ITS1 |
its1 |
CTTGGTCATTTAGAGGAAGTAA |
|
its2 |
GCTGCGTTCTTCATCGATGC |
||
|
ITS2 |
its3 |
GCATCGATGAAGAACGCAGC |
|
|
its4 |
TCCTCCGCTTATTGATATGC |
*注:只有两端完全测通的Reads (Tags)才能用于进一步的分析,目前短读长测序最长测序读长为PE300,片段范围在530以上的建议选择长读长测序来做。
信息分析内容
下机数据经过数据过滤,滤除低质量的reads,剩余高质量的Clean data方可用于后期分析;通过reads之间的Overlap关系将reads拼接成Tags;在给定的相似度下将Tags聚成OTU,然后通过OTU与数据库比对,对OTU进行物种注释;基于OTU和物种注释结果进行样品物种复杂度分析以及组间物种差异分析。
|
分析模块 |
信息分析条款 |
|
数据处理 |
数据过滤 |
|
Reads拼接 |
|
|
OTU聚类及分析 |
OTU统计 |
|
OTU venn图分析 |
|
|
Core-Pan OTU分析 |
|
|
OTU PCA分析 |
|
|
OTU NMDS分析 |
|
|
物种累计曲线分析 |
|
|
OTU PLS-DA分析 |
|
|
OTU Rank 曲线 |
|
|
物种分类 |
物种丰度柱状图(多方案展示) |
|
物种丰度热图(多方案) |
|
|
物种系统发育进化分析 |
|
|
GraPhlAn 物种组成图 |
|
|
物种PCA分析(样本≥ 3) |
|
|
菌群分型分析样本(肠道样本) |
|
|
单个样品多样性分析(alpha多样性) |
Alpha多样性统计表(包括observed species指数(sobs)、chao1指数、ace指数、shannon指数和simpson指数) |
|
Alpha多样性稀释曲线(多方案) |
|
|
Alpha多样性盒形图 |
|
|
样品间多样性比较分析(beta多样性) |
Beta多样性热图(Bray-Curtis,weighted UniFrac,unweighted UniFrac) |
|
样品聚类树 |
|
|
PCoA |
|
|
UPGMA 聚类树与丰度组合图 |
|
|
Beta多样性指数组间差异分析(分组≥2,每组样本数≥3) |
|
|
物种差异分析(分组≥2,每组样本数≥3) |
LEfSe分析 |
|
Wilcox Test |
|
|
Kruskal Test |
|
|
相似性分析检验分析(Analysis of similarity,ANOSIM) |
|
|
PERMANOVA/Adonis分析(如关注物种与表型的关联需提供表型信息,否则默认分析物种与分组关联) |
|
|
关键物种差异比较柱状图 |
|
|
功能分析 |
KEGG功能预测 |
|
KEGG对应酶的编号及通路图 |
|
|
COG功能预测 |
|
|
功能差异分析(Wilcox Test、STAMP) |
|
|
关联分析与模型预测(个性化分析) |
CCA/RDA分析(需提供详细的环境因子数据) |
|
Spearman相关系数分析(需提供表型信息) |
|
|
物种间相关系数网络图分析 |
|
|
随机森林--ROC曲线(分组=2,每组样本数≥30) |
|
|
SourceTracker(需要提供样本来源信息) |
案例一:女性生殖道微环境及其与生殖健康的关联
The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases (Nature communications, 2017).
该项研究是目前最大规模的育龄女性生殖道微生态研究项目,采用16S检测技术对女性盆腔与上下生殖道各部位菌群分布及与生殖系统疾病的关联进行研究。其研究成果打破了“盆腔和上生殖道为无菌环境”的传统认知,发现正常女性盆腔与上生殖道亦存在微生物,这首次揭示了生殖道从阴道到宫颈管、宫腔、输卵管,直至盆腔的菌群结构具有一定连续性,并提出盆腔与女性生殖道微生态环境和生殖系统健康及相关疾病具有重要的关联性。
样本来源:采集了110名因良性疾病接受手术治疗的育龄女性生殖道不同部位的样本,包括阴道下1/3(CL)、阴道后穹窿(CU)、宫颈管(CV)、子宫腔(ET)、输卵管(FL)及盆腔液(PF)
主要方法:16S rRNA基因扩增测序分析、实时定量PCR和微生物传统培养法分析。
主要分析点:物种分析、样本间多样性分析、功能预测、表型关联分析、评估模型构建。
主要结果:
- 上生殖道并非无菌环境,从阴道到输卵管及盆腔,各部位微生物物种组成呈解剖结构上的渐进性变化
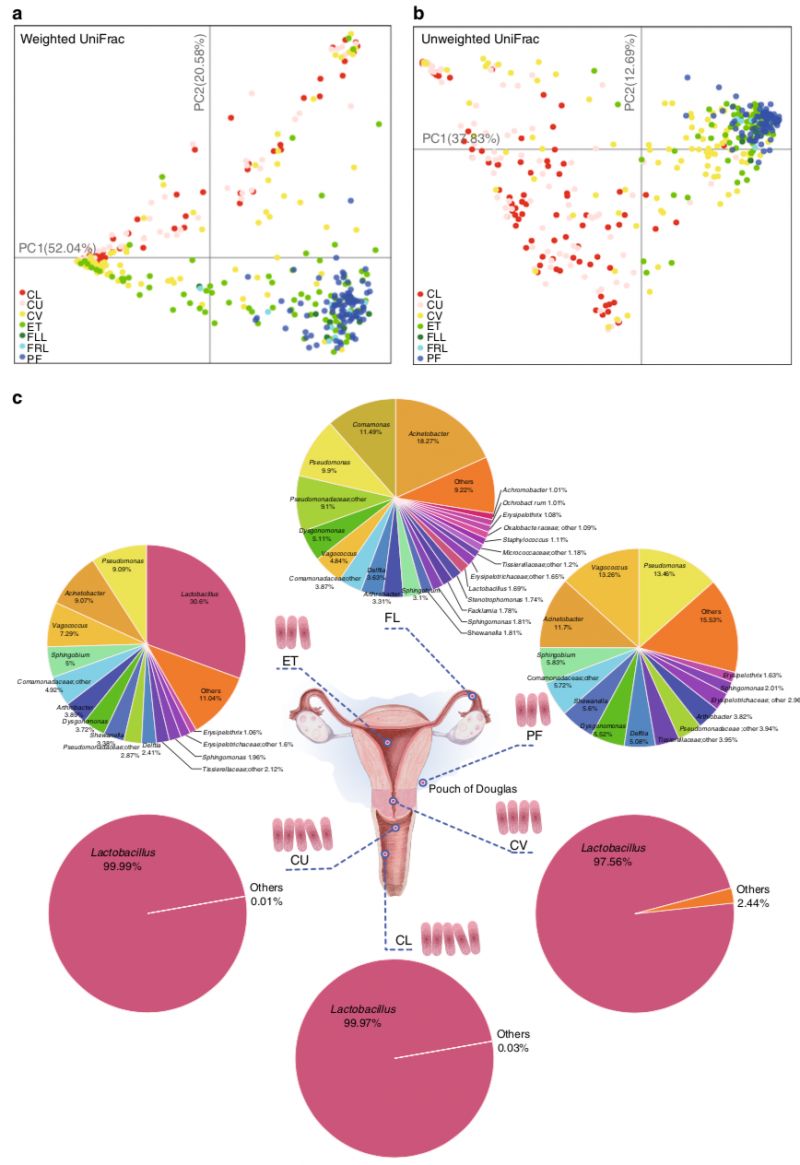
图1. 生殖道不同部位微生物群落结构。a, Weighted UniFrac PCoA图;b, Unweighted UniFrac PCoA图;c, 生殖道不同部位微生物丰度饼图(属水平)。
2. 同一个体不同部位的样本间具有很高的相关性,不同个体间菌群组成变化明显。此外,同一个体的宫颈管样本与腹腔液样本具有显著的相关性,表明在普通人群中可以通过分析易取得的宫颈粘液样本来评价宫腔和腹腔的菌群分布状况。
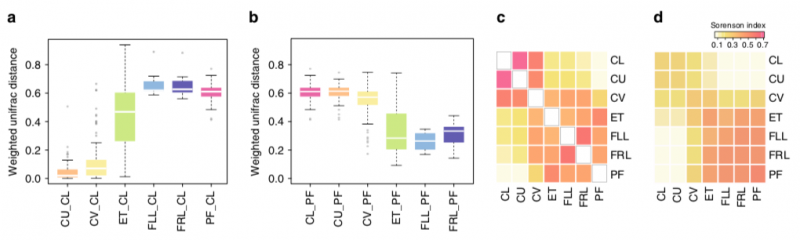
图2. 不同个体/同一个体不同部位微生物群落结构一致性。a, 同一个体不同部位的微生物群落结构与阴道下1/3(CL)的相似性;b, 同一个体不同部位的微生物群落结构与盆腔液(PF)的相似性;c, 同一个体不同部位的微生物群落结构Sorenson indices结果;d, 不同个体不同部位的微生物群落结构Sorenson indices结果
3. 对表型信息进行关联分析发现月经周期、孕产次数以及子宫内膜异位症、子宫腺肌症等多种疾病相关的不孕等都与生殖道菌群的变化有关。
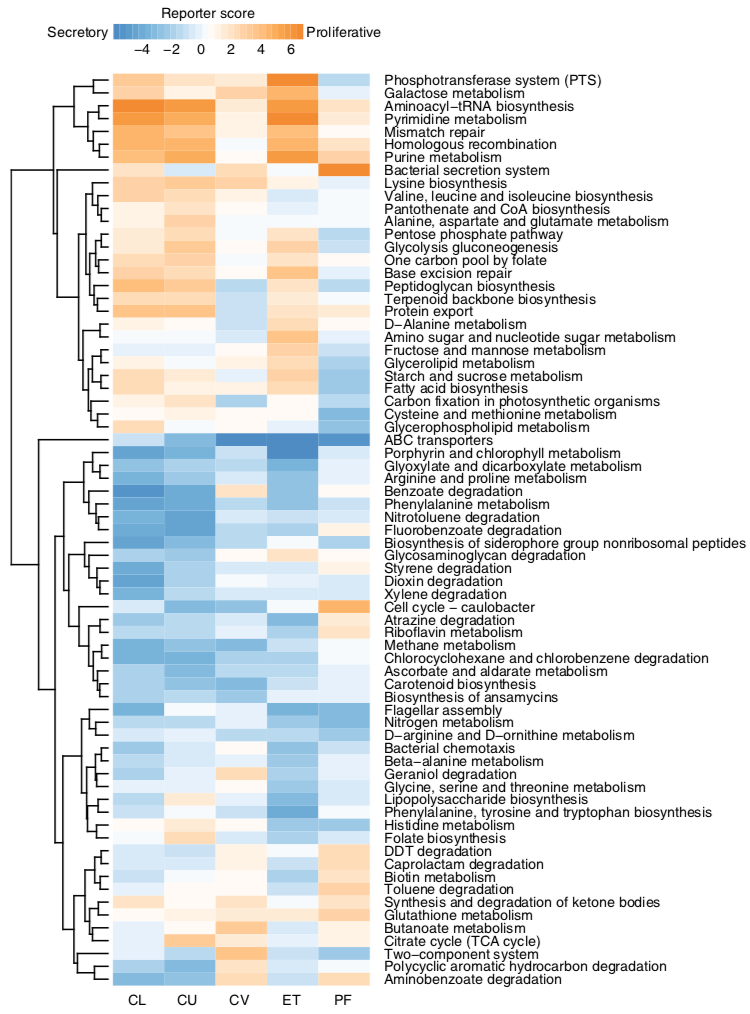
图3. 生殖道微生物功能(预测的)与女性生理周期的关联分析
案例二:猪肠道菌群衍生物细菌素可增强早期断奶仔猪腹泻抵抗力
A Microbiota-Derived Bacteriocin Targets the Host to Confer Diarrhea Resistance in Early-Weaned Piglets (Cell host & microbe, 2018).
在猪养殖过程中,早期断奶可以缩短猪的屠宰周期并改善母猪的繁殖性能。然而早期断奶容易导致应激性腹泻,仔猪死亡率上升,生长性能降低。使用抗生素可以预防仔猪断奶腹泻,降低饲养成本,但是由于病原菌抗生素抗性和抗生素残留问题,欧盟已完全禁止在动物饲养中使用抗生素。因此,寻找抗生素替代品以预防早期断奶仔猪的腹泻对于畜牧业和粮食安全至关重要。哺乳动物肠道菌群与宿主健康密切相关,通过粪菌移植或益生菌/益生元调控肠道菌群已成为有前景的胃肠道疾病治疗策略。
与商业杂交LY仔猪相比,CM仔猪(中国本土品种)对早期断奶应激诱导的腹泻抵抗力更强。本研究在早期断奶之前给LY仔猪口服CM仔猪粪便微生物群, LY仔猪的腹泻抗性增强。通过比较粪菌移植组和对照组LY仔猪肠道微生物群的相对丰度,鉴定到两个可能跟腹泻抗性相关的菌种加氏乳杆菌LA39(Lactobacillus gasseri LA39 )和乳酸杆菌(Lacto-bacillus frumenti),并通过qPCR进行验证。腹泻抵抗依赖于细菌素gassericin A,gassericin A与角蛋白19(KRT19)在肠上皮细胞质膜上的结合对于增强液体吸收和减少分泌至关重要。本研究结果表明L. gasseri LA39和L. frumenti可能是预防哺乳动物腹泻的有效抗生素替代品。
方案设计:
对LY仔猪和CM仔猪按不同处理进行分组如下:
①LY: LY仔猪,不经任何处理,n=3;
②LY (saline):LY仔猪,day10-day18隔日口服生理盐水,n=3
③LY (high dose):LY仔猪,day10-day18隔日口服高浓度CM仔猪粪菌悬液,n=3;
④LY (low dose): LY仔猪,day10-day18隔日口服低浓度CM仔猪粪菌悬液,n=3;
⑤LY (oxytetracycline) :LY仔猪,断奶日(day21)肌肉注射长效土霉素,n=3
⑥CM:CM仔猪,不经任何处理,n=3
以上各组仔猪在断奶后第3,5,6,8,11天收集粪便样本,进行16S V4和ITS2测序。
主要结果:
1. 粪菌移植仔猪腹泻症状缓解,肠道菌群结构和功能发生改变
1) 接受CM粪菌移植的LY仔猪早期断奶症状缓解
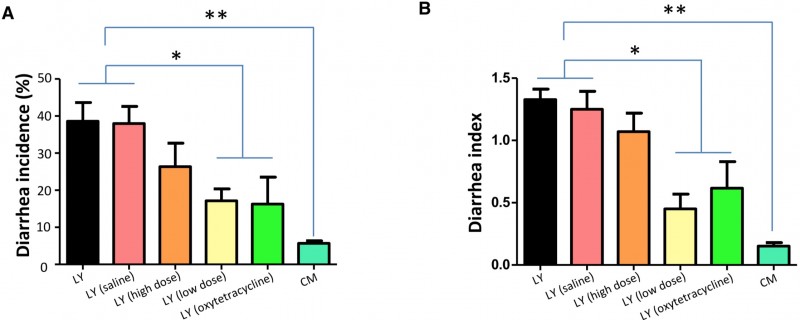
图1. 各组仔猪腹泻发病率(A)和腹泻指数(B)
2) LY (saline)组,LY (low dose)组和CM组粪便细菌有明显差异;真菌无明显分群。
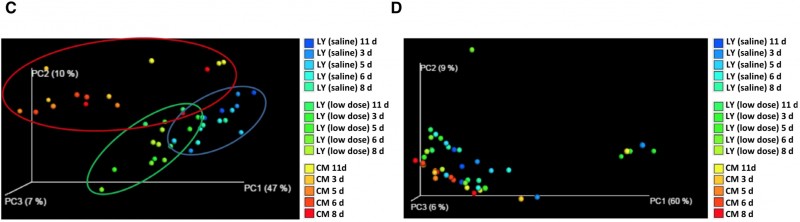
图2. 细菌和真菌群落PCoA 结果
3) 接受CM粪菌移植的LY仔猪细菌/真菌群落多样性发生了改变
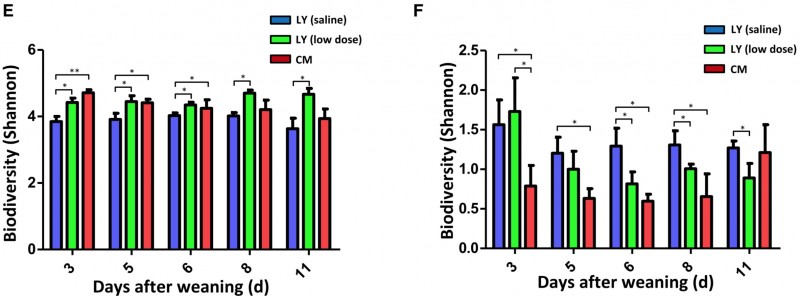
图3. 细菌和真菌alpha diversity 结果
4) LY (saline)组,LY (low dose)组和CM组粪便细菌功能结构有明显差异;
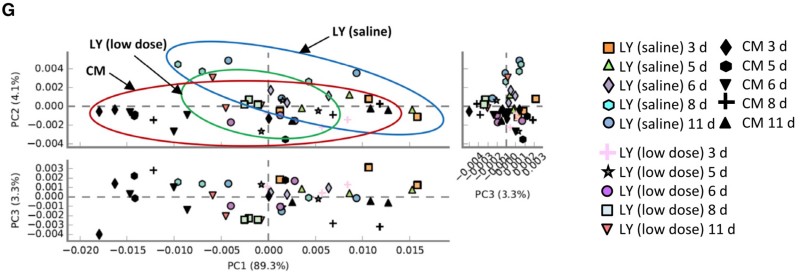
图4. 仔猪肠道菌群功能pCoA图(基于PICRUSt软件 KEGG pathway分析)
2. 口服腹泻抗性相关的肠道微生物可预防仔猪早期断奶应激引起的腹泻。鉴定得到5个跟腹泻抗性相关的菌种,其中加氏乳杆菌LA39(Lactobacillus gasseri LA39)和乳酸杆菌(Lactobacillus frumenti)单独服用即可增强仔猪腹泻抗性。
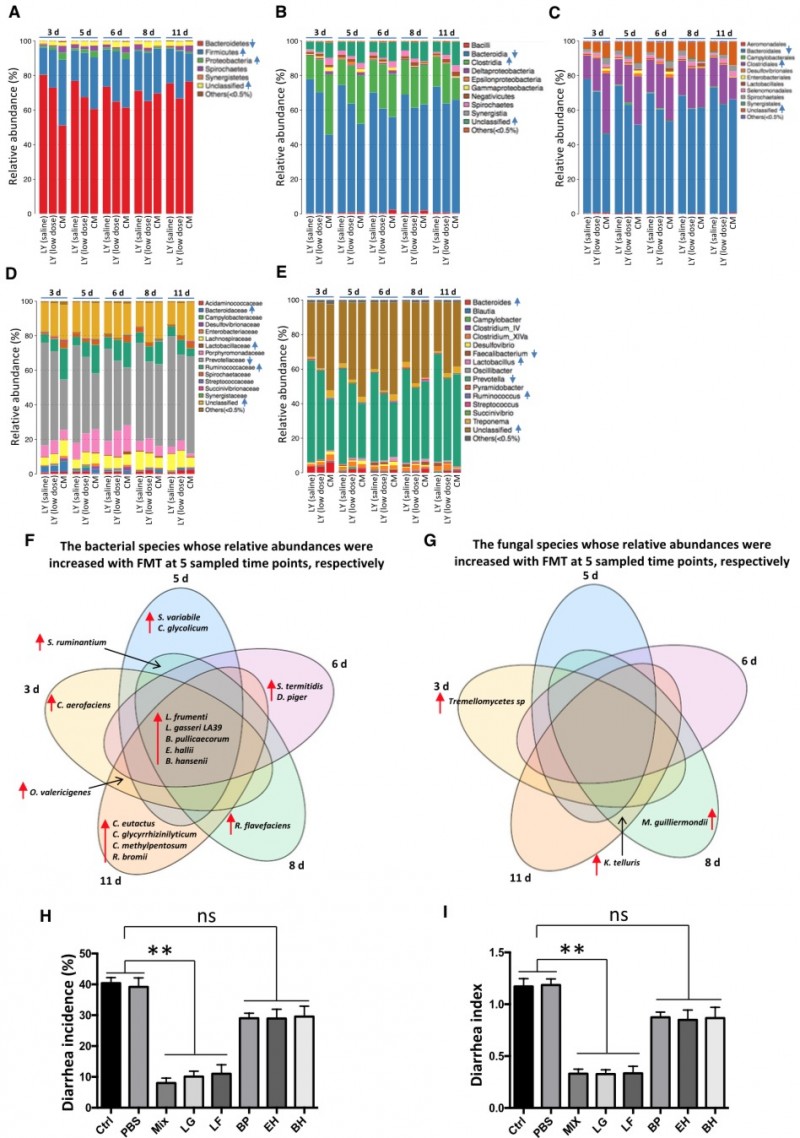
图5. A-E, 三组仔猪肠道菌群物种注释结果(门、纲、目、科、属);F,G,粪菌移植仔猪肠道菌群变化物种韦恩图(F,细菌;G,真菌);H,I,特定菌株对仔猪腹泻发生率(H)和腹泻抗性(I)的影响。
3. 肠道微生物介导的抗腹泻功能依赖于分泌型Gassericin A。混合菌群移植和单独服用LG/LF菌株的LY仔猪肠道菌群Gassericin A编码基因GaaA丰度升高;口服GaaA缺陷LG菌株不能改善LY仔猪腹泻症状。
4. Gassericin A与肠上皮细胞中的细胞质膜结合,增强肠液吸收,减少肠液分泌。
5. KRT19介导的Gassericin A与细胞质膜结合,对于Gassericin A介导的肠液吸收增强和液体分泌减少至关重要。
Gassericin A通过激活由雷帕霉素机制靶标介导的磷酸二酯酶活性降低细胞周期核苷酸水平,增加肠液吸收并减少肠液分泌。
DNA样本送样建议
|
样本类型 |
总量 |
浓度 |
完整性(胶图) |
纯度 |
|
|
扩增子建库 |
基因组DNA |
≥0ng(推荐50ng以上) |
≥0ng/μl |
必须为基因组样本 |
无蛋白,RNA/盐离子等污染,样本无色透明不粘稠 |
|
PCR-Free建库 |
PCR 产物 |
≥3μg |
≥30ng/μL |
条带清晰无弥散 |
|
Meta rDNA Amplicon Sequencing包括Meta rDNA V3,V6,V4,V1-V3,V3-V4,V4-V5,V5-V6,ITS1,ITS2区域Amplicon建库,这类文库主要核心实验环节是PCR扩增,扩增成功率受模板DNA和体系纯度(含有杂质,盐离子,色素,腐殖酸等)等因素影响,所以Meta rDNA Amplicon需要根据第一次建库扩增成功与否来判断样品是否合格。
组织样本送样建议
|
组织类型 |
Meta扩增子测序 |
|
粪便样本 |
≥100mg |
|
土壤样本 |
≥200mg |
常见样本采样建议
(一)液氮速冻法
① 人和大型动物粪便样品的采集
a) 准备好便盆和粪便容器,洗手,带上手套收集新鲜的粪便样本;
b) 在实验室将装好受试者粪便样本,立即进行分装并标记;
c) 用无菌牙签或粪便取样器截取样品中段里部(粪便表层含有肠粘膜脱落细胞;外部容易污染,且接触空气后,部分细菌DNA 开始降解),取约50~100mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取3-5管备份
d) 分装好后,立即液氮速冻或直接放入-80℃低温保存,送样时选择干冰运输寄送。
如粪便样品量较多或不能马上冻存,最迟要在2小时之内全部收集完。
② 小型动物(如鼠)颗粒粪便样品采集
动物颗粒粪便样品,动物排便后立即装入2.0mL离心管中(小鼠粪便3粒每管),放入-80℃低温保存,送样时选择干冰运输寄送。
③ 肠道内容物样品采集
a) 用无菌解剖刀,在无菌状态下取出整个肠道,切取所需肠段的内容物(条件允许的话,可在无菌操作台进行);
b) 用无菌手术刀挖取内容物,装入无菌2.0mL离心管中,每管取约50~100mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取3-5管备份;
c) 分装好后,立即放入-80℃低温保存,送样时选择干冰运输寄送。
④ 肠道组织样本采集
a) 组织样本用无菌磷酸盐缓冲液轻轻清洗,直到没有内容物流出;
b) 用无菌的显微镜玻片刮取附着在表面的组织细菌,转移到无菌的2.0mL离心管中;
c) 立即转入-80℃低温保存,送样时选择干冰运输寄送。
⑤ 土壤meta样品采集
a) 根据研究目的确定采样范围,取样器具要事先消毒灭菌处理,开始采样;
b) 去除表面浮土,使用乙醇火烧的铲子挖取地下5~20cm的土层;
c) 去除可见杂质后,土壤过2mm筛网,建议每个样品从3个及以上采样点采集并混合而成,把土样装入无菌2.0mL离心管中,每管取约50~100mg (约花生米大小,装入2.0mL离心管不超过1/3体积)到无菌的2.0mL离心管中,每个样本取3-5管备份;
d) 分装好后,立即转入-80℃低温保存,送样时选择干冰运输寄送。
⑥ 水体样本采集
a) 根据研究目的确定采样深度和范围;
b) 采集好的水样需要通过滤膜进行过滤,可以根据水样的浑浊程度选择相应孔径的滤膜;
c) 将滤膜转移到2.0mL离心管中,立即转移至-80℃低温保存,送样时选择干冰运输寄送。
清亮水样:可选择小孔径的滤膜,一般选0.22μm或0.45μm的滤膜,过滤水样体积大于10L;
浑浊水样:过滤前静置分离悬浮颗粒,也可以用大孔径的滤膜预过滤一遍,再用小孔径的滤膜进行过滤。
(二)商业核酸保护液保存法
人粪便样品可采用常温采样套装,具体请按照说明书操作保存运输样品。
Q1. 16S产品有样品数量的要求,需要所有样品准备好了才能进行测序分析吗?
A1:从分析角度,同一处理建议至少4个样本进行分析。从科学的角度来讲,最好能够整批样品同时测序分析,既可以减少不同批次间的系统误差,还能节省项目周期若样品准备有困难。也可以分批次启动,但可能带来系统误差问题。
Q2. 不同环境样本数据量要求?
A2:一般推荐简单环境(如人肠道、发酵液等)测序数据量为50,000tags;复杂环境(如土壤、海水)等推荐数据量为100,000tags以上。
Q3. 16S测序能不能进行功能分析?
A3:16S测序主要是基于16S rDNA 序列相似性进行OTU聚类进而进行物种注释及相关多样性研究。因为16S测序并没有测到对应的基因组信息,不能直接基于测序结果进行功能注释。
利用软件PICRUSt可以进行16S功能预测,该软件的原理是:对由16S测序分析得到的OTU丰度进行拷贝数均⼀化,得到样品中可能出现的细菌及数目,从细菌的基因组信息得到对应的基因信息及注释信息,再结合均⼀化的OTU丰度来预测样品中可能存在的各级KEGG通路及丰度值以及COG功能信息及丰度值。
基于16S的功能预测可以作为后续功能研究提供参考,但由于该分析不能反映群落中因基因表达差异导致的功能差异。如果主要关注功能差异,最好选择宏基因组测序来进行功能研究。
Q4. 16S相关文章中选择的测序区域各不相同(如V4,V3-V4,V1-V3,V3等),选择的依据是什么,选择哪个区域比较好?
A4:不同物种不同区域多样性不同,选择不同区域测序结果会有不同,可能会造成物种多样性的低估或高估。在非全长16S测序的情况下,测序区域也并非越长越好,跟全长16S结果最相近的测序区域即是最优选择。
如无特殊要求,我们一般推荐V4区测序,有文献表明V4区测序结果和全长的结果一致性较好。
根据我们大量的项目经验,目前测序项目较多的区域为V4, V1-V3, V3-V4, V4-V5, V5-V6,具体项目测序区域也可参考相关文献进行选择。
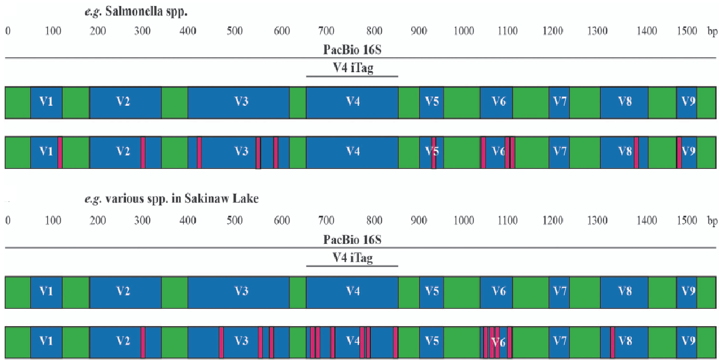
图1. 不同物种16S各可变区变异程度不同
Q5. 16S样本要求有哪些,样本准备有哪些注意事项?
A5:对16S样本要求如下:
|
Meta扩增子测序 |
|||||
|
样本类型 |
总量 |
浓度 |
完整性(胶图) |
纯度 |
|
|
Meta rDNA
Amplicon |
Genomic DNA |
≥0ng(推荐50ng以上) |
≥0ng/μl |
必须为基因组样本 |
无蛋白,RNA/盐离子等污染,样本无色透明不粘稠 |
|
Meta rDNA
Amplicon PCR-free Library |
PCR products |
≥3μg |
≥30ng/μL |
条带清晰无弥散 |
|
一般16S建库选择基因组样本,推荐样本量在50ng以上,如果样本准备困难,大于0ng也可以尝试建库。
16S产品建库受模板DNA和体系纯度(含有杂质,盐离子,色素,腐殖酸等)等因素影响,在取样过程中,尽量减少宿主细胞含量及其他杂质的影响。
样本采集后尽快放入-80℃冻存,干冰运输,减少样本降解导致的微生物群落结构变化。
Q6. 我的样本检测合格了,样本量也达到了推荐样本量要求,却建库失败了,可能是什么原因造成的,有没有什么解决方案?
A6:这种情况常见于宿主微生物样本,该类样本通常还有大量的宿主DNA,由于样本检测中不能区分宿主和微生物DNA,实际检测到的DNA其实绝大多数都是宿主DNA,导致检测到的样本量很高,却建库失败的情况。
对于这类样本,推荐在样本制备过程中进行特殊处理,尽量减少宿主DNA含量。目前市面上有可以去宿主DNA的试剂盒(如QIAamp DNA Microbiome Kit),对于宿主含量较高的样本如唾液、粘膜样本等,可以选择对应的试剂盒处理。
Q7. 16S测序一般推荐多少样本量?
A7:16S样本多样性及组间差异分析是基于统计结果进行的分析,一般样本数越多,统计结果越准确。最低样本数要求如下:
样本间多样性分析(n≥4),组间多样性分析样本(组别≥2,每组样本数n≥3)。
Q8. 16S测序如果样本含有较多宿主DNA,是否需要增加测序数据量?
A8:16S扩增子测序是对微生物16S rDNA 特定区域进行扩增并测序分析的产品,扩增产物不包含宿主的信息。所以样本中宿主DNA并不会对测序结果造成影响(宿主DNA含量过高可能会导致建库失败),测序过程中也无需增加数据量。
宏基因组测序是对环境中所有物种进行全基因组测序的产品,样本中宿主DNA含量较高会导致宏基因组测序宿主比例偏高,微生物有效数据量相对较少,这种情况下可以通过增加测序数据量来增加有效数据。