- 首页 > 蛋白磷酸化修饰分析
蛋白磷酸化修饰分析
蛋白翻译后修饰(Post-translational modification, PTM)指蛋白经生物合成后的共价修饰及酶学修饰,修饰作用可发生在蛋白氨基酸侧链及其C或N末端,通过对现有20种氨基酸官能团的改造或者引入新的基团(如磷酸基、乙酰基等)来实现其功能的扩展,大多数真核生物所表达的蛋白质需经过一系列的翻译后加工和修饰才能形成最终复杂的功能执行体,因此蛋白质的翻译后修饰成为蛋白质组学研究的重要方面。
翻译后修饰蛋白质在样本中含量低且动态范围广,其相关研究极具挑战性,亲和富集、多维分离等技术与生物质谱的结合为翻译后修饰蛋白质组学的发展提供了契机。目前,已进行规模化研究的蛋白修饰主要有磷酸化(Phosphorylation)、乙酰化(Acetylation)、糖基化(Glycosylation)、泛素化(Ubiquitination)等。
其中,蛋白质磷酸化(Phosphorylation)是最常见、最重要的一种蛋白翻译后修饰方式,发生在蛋白质丝氨酸、苏氨酸以及酪氨酸上,它由两个作用相反的酶系——蛋白激酶和磷酸酶进行调控。
蛋白磷酸化修饰分析技术,主要包括蛋白磷酸化鉴定(纯化蛋白磷酸化鉴定/磷酸化蛋白全谱鉴定)和蛋白磷酸化定量。华大基因的蛋白磷酸化定量产品主要包括以下几种。
|
产品名称 |
产品优势 |
|
蛋白磷酸化修饰iTRAQ/IBT/TMT标记定量 |
一次性比较多个样本,极大地降低了实验误差,提高定量准确性。 |
|
蛋白磷酸化修饰 4D-DIA非标记定量 |
高深度:1h鉴定磷酸化肽段近15,000;高准确性:定量相关性达0.95;多维高精度:同4D-DIA常规蛋白定量强强联合,探索蛋白表达“全景图”。 |
|
蛋白磷酸化修饰 PRM靶向定量 |
采用“full MS+PRM”;PRM list导出成功率高;操作便捷稳定,省去碰撞能量(CE)优化;定量更准确,含更丰富的母离子和子离子信息辅助定量;质控严格,定量时样品间内参磷酸肽的CV小于25%。 |
(1)蛋白磷酸化修饰标记定量
蛋白磷酸化修饰标记定量是基于高精度、高灵敏度质谱仪,将iTRAQ/IBT/TMT定量技术与TiO2富集技术相结合,对生物体的修饰蛋白进行鉴定并相对定量,从而达到研究生物体内修饰蛋白质组动态变化与集体表型改变之间关系的目的。
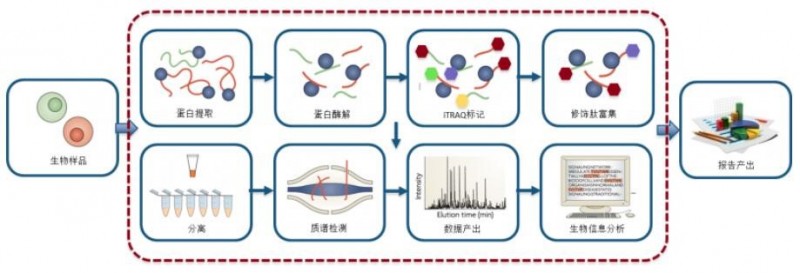
图1 蛋白磷酸化修饰iTRAQ定量技术路线
(2)蛋白磷酸化修饰4D-DIA非标记定量
基于DIA全景扫描的无偏模式,DIA磷酸化蛋白质组得以突破传统DDA的限制,能够有效解决磷酸化修饰定性定量的难题,推进磷酸化分析向更高鉴定深度、更高定量重复性和定量准确性的方向发展。4D技术在原有保留时间、质荷比和离子强度的基础上,增加了离子淌度(CCS),在检测过程中能够进一步降低待测样本的复杂程度,显著提高了对蛋白修饰的检测能力,4D技术加持下的DIA磷酸化蛋白组技术,实现了对磷酸化蛋白的超高深度鉴定和超精准定量。
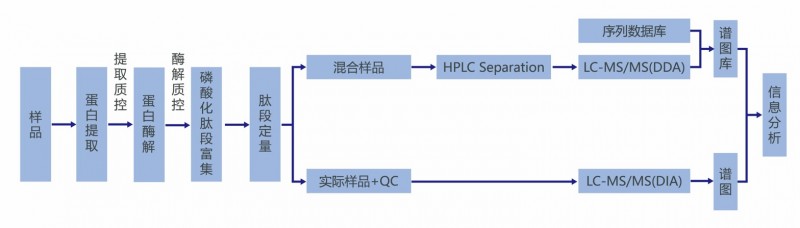
图2 蛋白磷酸化修饰4D-DIA定量技术路线
(3)蛋白磷酸化修饰PRM靶向定量分析
PRM(parallel reaction monitoring),平行反应监测技术,通过串联质谱,有目的地筛选目标蛋白肽段进行定量的靶向蛋白质组研究策略之一。PRM和MRM(multiple reaction monitoring,或SRM,selected reaction monitoring)技术类似,都可作为目标蛋白定量研究策略,需要先通过一级质谱将目标蛋白的肽段筛选出来;不同的是,在二级质谱时,PRM会将目标肽段所有碎裂的子离子信号进行采集,而MRM只采集筛选到的特定子离子信号,通过液质联用(LC-MS/MS)的方式,主要对感兴趣的磷酸化修饰蛋白进行定量验证。
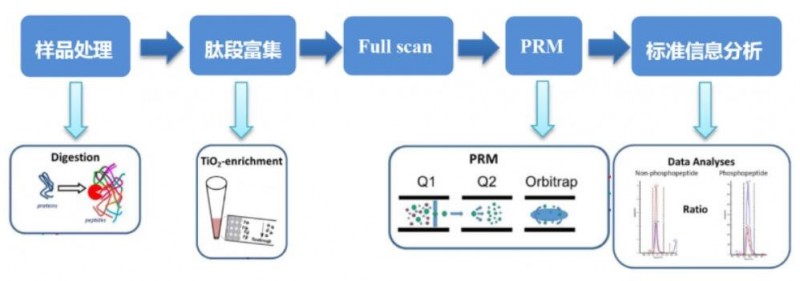
图3 蛋白磷酸化修饰PRM靶向定量技术路线
信息分析内容(以4D-DIA磷酸化为例)
|
信息分析条款 |
信息分析内容 |
|
标准信息分析 |
1 项目概述及质控分析 1.1项目概述 1.2数据质控 1.3鉴定与定量细节表 2 DDA谱图库 3 磷酸化蛋白组鉴定 3.1磷酸化蛋白组鉴定结果统计 3.2磷酸化蛋白质组鉴定的质量评估 3.3磷酸化位点统计 4 磷酸化蛋白组定量 4.1差异定量分析 4.2主成分分析 4.3聚类分析 4.4时间序列分析 5 磷酸化蛋白组功能注释 5.1磷酸化蛋白质GO注释 5.2磷酸化蛋白质KOG注释 5.3磷酸化蛋白质Pathway代谢通路注释 6 差异磷酸化肽段对应蛋白的功能注释 6.1 GO富集分析 6.2 Pathway富集分析 6.3 KOG注释 6.4差异磷酸化肽段对应蛋白互作分析 6.5差异磷酸化肽段对应蛋白的亚细胞定位 7 磷酸化肽段Motif分析 |
|
定制化信息分析 |
可结合客户的需求,协商确定定制化信息分析服务内容。 |
1、基于数据非依赖型(DIA)质谱采集的方法的快速磷酸化蛋白质组学
Bekker-Jensen D B, Bernhardt O M, Hogrebe A, et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries[. Nature Communications, 2020.
背景:
磷酸化是最重要的翻译后修饰之一,目前大多数磷酸化蛋白质组学研究需要通过质谱技术进行数天甚至数周的数据采集和分析,分析大量样品中的磷酸化位点仍具有挑战性。
实验设计:
通过开发一种优化的DIA磷酸化蛋白质组学方法,系统地、可重复地分析数百个样品中的磷酸化位点。同时运用此技术来确定表皮生长因子信号通路的主要蛋白激酶的磷酸化位点。
主要结果:
1)优化了分析流程,通过调整参数设置优化仪器设置从而获得最佳DIA性能。与目前先进的基于数据依赖采集模式(DDA)的磷酸化蛋白组学相比,基于DIA的磷酸化蛋白组学具有更宽的动态范围、更高的鉴定重复性、更高的定量灵敏度和准确性。
2)以视网膜色素上皮细胞(Retinal pigment epithelium, RPE1)为实验对象,使用不同MEK激酶抑制剂(MEK kinase inhibitors)与RPE1细胞反应以诱导产生相应的信号通路。采用DDA和DIA方法采集的数据通仍然通过MQ软件与SQ软件进行分析,DIA的整体结果均优于DDA结果。
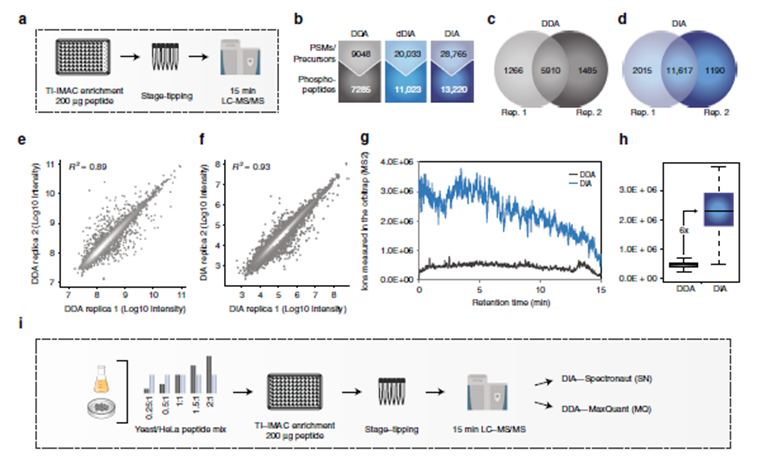
图1 DDA和DIA磷酸化蛋白质组学鉴定和定量
2、草鱼柔软和坚硬肌肉的磷酸化蛋白组学分析
Quantitative phosphoproteomic analysis of soft and firm grass carp muscle. Food Chemistry, 2020
背景:鱼的肌肉硬度是消费者接受的一个重要质量特征,也是鱼产品生产者、加工商和消费者重要的质量指标之一,更好的质地使鱼易于加工成高质量的产品。磷酸化蛋白组学分析被证明是一种了解与肉质形成相关的生物过程的有效方法,如肉色稳定性、pH值下降和肌肉糖酵解,但这项技术很少应用于鱼类。
实验设计:
6个脆草鱼和6个普通草鱼的肌肉组织进行磷酸化iTRAQ。
主要结果:
在脆草鱼中,鉴定到27个上调和22个下调的磷酸化肽段及其潜在的上游激酶。蛋白互作分析将这些磷酸化蛋白聚类为四组:12个骨骼肌纤维蛋白、1个结缔组织蛋白、3个信号调节蛋白和4个碳水化合物代谢蛋白;肌球蛋白、肌动蛋白、原肌球蛋白、紧密连接途径和糖酵解途径可能有助于增强肌肉硬度。这些结果为蛋白质磷酸化在鱼类肌肉硬度中的作用提供了新的见解,并将有助于提高鱼产品的质量。
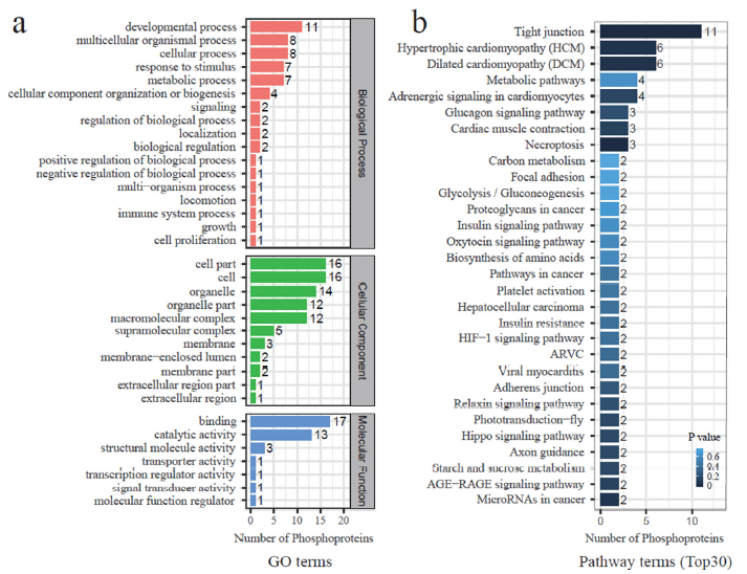
图2 脆皮草鱼和普通草鱼肌肉中差异磷酸化蛋白的GO和pathway富集分析
a. GO富集分析; b. KEGG富集分析(Top30)
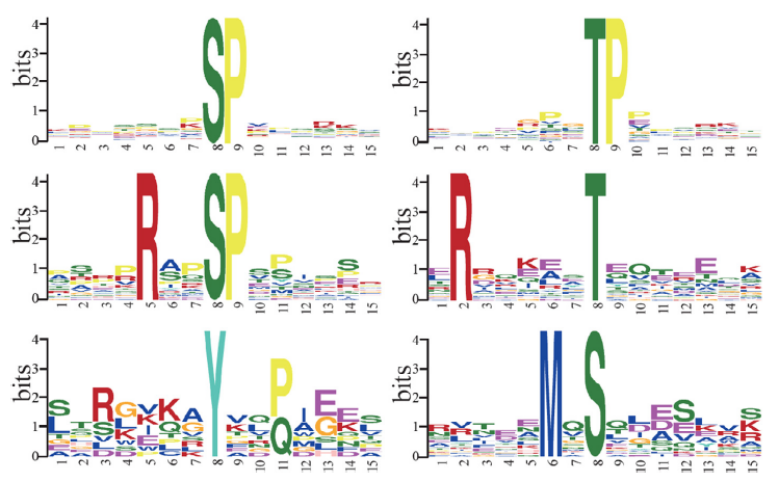
图3 高度表示它们在各自位置出现的频率,颜色代表了它们的理化性质
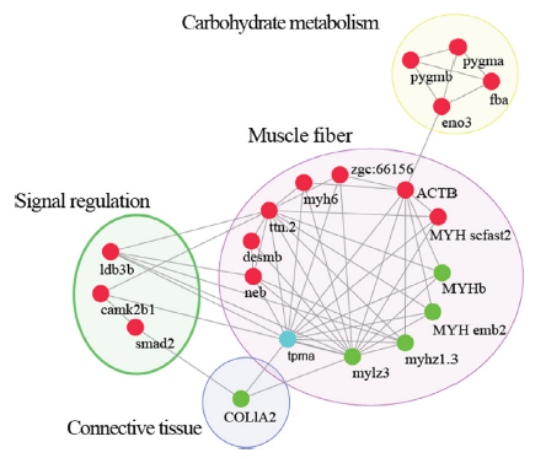
图4 磷酸化蛋白质-蛋白质相互作用网络图
红色代表上调,绿色代表下调,蓝色代表上调和下调同时存在
1、项目概述及质控分析
基于磷酸化蛋白质组定量分析结果,分别从磷酸化肽段、磷酸化位点和磷酸化蛋白三个层面统计整体项目及各样本的鉴定情况。
之后分别从组内变异系数,主成分分析以及样本定量相关性等方面评估数据的质量。当样本数目较多时,会在连续上机样本中间断插入QC样本,从而能在质控分析中评估实验条件,以便确保实验的稳定性和重复性。
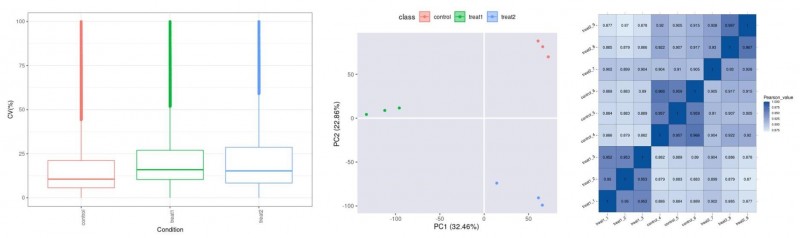
图1 CV分布(左)、主成分分析(中)和样本相关性分析热图(右)
2、磷酸化肽段定量及差异磷酸化蛋白富集分析
在4D-DIA定量中,通常通过差异倍数大于1.5或小于2/3,同时满足统计检验P-value值小于0.05筛选显著差异表达磷酸化肽段,并绘制火山图。对差异磷酸化肽段对应的蛋白质,分别进行GO、KOG和KEGG pathway富集等多种功能注释富集分析。
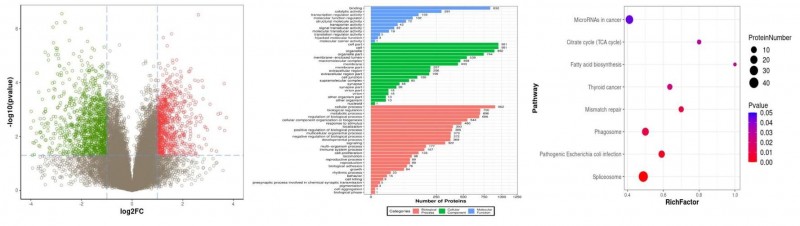
图2 磷酸化肽段定量及差异磷酸化蛋白富集分析
左图为磷酸化肽段定量分析火山图,横轴为肽段定量值(log2化),纵轴为统计检验的P-value(-log化);分布在该火山图左上、右上两区域的肽段为显著差异的肽段、中图为差异磷酸化蛋白GO富集分析,右图为差异磷酸化蛋白KEGG Pathway富集分析气泡图。
3、磷酸化位点motif分析和激酶-底物分析
Motif是比较有特征的短序列,会多次出现并被假设拥有生物学功能。而且,经常是一些具有序列特异性的蛋白结合位点(如转录因子)或者涉及到重要生物过程。

图3 蛋白磷酸化位点Motif分析
人基因组注释信息显示人类有518个激酶,主要分为9个组:AGC(蛋白激酶A、G、C组)、Atypical(非典型组)、CAMK(钙调节激酶组)、CK1(酪蛋白激酶组)、CMGC(蛋白激酶CDK、MAPK、GSK3、CLK组)、STE(酵母STE7、11、20同源基因激酶组)、TK(酪氨酸激酶组)、TKL(类酪氨酸激酶组)和其他,90个家族、145个亚族。不同家族偏向有不一样的功能。
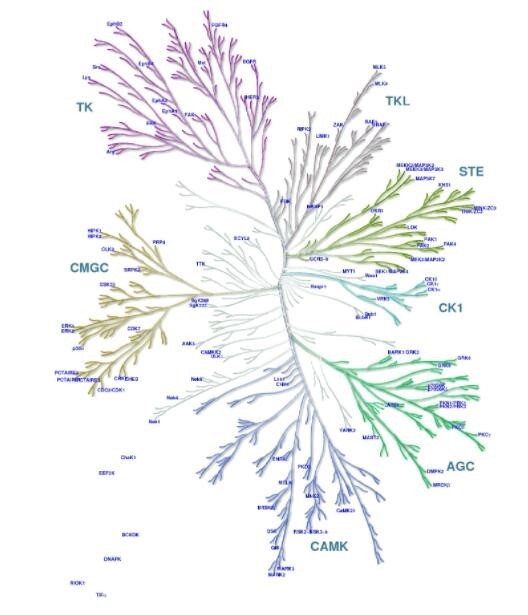
图4 激酶-底物分析
|
样品类型 |
iTRAQ/IBT/TMT/ /PRM 磷酸化蛋白定量 |
4D-DIA/4D label-free 磷酸化蛋白定量 |
DIA磷酸化蛋白定量 |
备注 |
|
|
动物类 |
常见动物组织:动物内脏(心、肝、脾、肺、肾)、皮肤、肌肉、脑等 |
≥20mg |
≥20mg |
≥100mg |
|
|
软体动物类(弓形虫、血吸虫、果蝇、螨虫、小菜蛾、灰飞虱、绦虫、蝉、涡鞭虫等) |
≥20mg |
≥20mg |
≥200mg |
|
|
|
细胞类 |
悬浮细胞、贴壁细胞 |
≥2×107 |
≥2×107 |
≥1×108 |
|
|
植物类 |
植物的嫩枝部(叶芽、嫩叶片)、藻类 |
≥2g |
≥2g |
≥5g |
|
|
植物的老叶、根、茎,树皮 |
≥4g |
≥4g |
≥8g |
|
|
|
植物的花蕾、花粉 |
≥200mg |
≥200mg |
≥2g |
|
|
|
植物的种子(水稻/小麦种子等)、果实(苹果、水蜜桃、梨) |
≥500mg |
≥2g |
≥2g |
|
|
|
微生物类 |
原核细菌(大肠杆菌、沃氏葡萄球菌等),真菌(酵母等) |
菌体≥200mg 或细胞数≥2×107 |
菌体≥200mg 或细胞数≥2×107 |
菌体≥500mg 或细胞数≥2×107 |
原核样品不推荐 做磷酸化修饰 |
|
蛋白液类 |
复杂样品蛋白液、蛋白粉末 |
≥1mg 浓度≥0.5μg/μL |
≥2mg 浓度≥0.5μg/μL |
≥20mg 浓度≥0.5μg/μL |
|
Q1:磷酸化位点是如何确定的?
A1:一种特定的翻译后修饰通常会作用于一定的氨基酸,经修饰后,这一类氨基酸会增加相同的分子量,如,磷酸化肽段因加入磷酸化基团而产生+80的质量偏移。基于质量偏移的理论,利用生物信息学方法可以分析质谱数据鉴定磷酸化翻译后修饰。
Q2:磷酸化蛋白质组富集策略有哪些?如何选择?
A2: 对于磷酸化蛋白的分离富集
1)抗体富集法:就是用识别磷酸化氨基酸残基的特异抗体进行免疫共沉淀,从复杂混合物中免疫沉淀目标蛋白质,是较为简单的方法;
2)激酶特异富集法:利用小分子的磷酸激酶抑制剂对其进行特异亲和层析成为激酶磷酸化蛋白质组学研究的重要方法;
3)亲和富集法:利用磷酸基团与一些金属离子或金属氧化物等的亲和作用,实现对磷酸化蛋白的富集。
对于磷酸化肽段的分离富集
1)化学修饰富集法:用亲和试剂取代磷酸化肽段上的磷酸集团, 再用亲和提取的方法从混合肽段中分离磷酸化肽段, 是一种有效的化学修饰法;
2)色谱分离富集法:包括IMAC富集法、金属氧化物(如TiO2等)和金属氢氧化物富集方法、阴阳离子交换富集法以及亲水性相互作用色谱法等;
3)MALDI靶盘富集法:为了满足对磷酸化蛋白的高通量分析以及微量样品的分析, 可以采用在线富集后直接质谱鉴定的方法。利用MALDI靶盘可实现直接在线富集检测。
据文献报道酪氨酸磷酸化比例最少,仅占整体磷酸化肽段的1%,如果实验目的只关注酪氨酸磷酸化的变化时,建议选择酪氨酸磷酸化抗体进行富集。如果实验关注的是非酪氨酸,或者是所有可以发生磷酸化的氨基酸(丝氨酸、苏氨酸、酪氨酸),由于抗丝、苏氨酸磷酸化抗体抗原决定簇较小,使得抗原抗体的结合位点村长空间障碍,特异性较差,所以建议选择IMAC或TiO2富集。
Q3:华大科技用于磷酸化富集的方法是什么?
A3:华大科技选用的是TiO2富集法,它是目前较为成熟的金属氧化物磷酸化肽富集法TiO2在不同的pH值下,可以表现为路易斯酸或路易斯碱。在酸性条件下,钛原子带正电表现为路易斯酸,可以与阴离子结合;在碱性条件下,则表现为路易斯碱,可以与阳离子结合。这样磷酸化肽段的磷酸基在酸性条件下与之结合,碱性条件下洗脱,达到富集磷酸化肽段的目的。
Q4:4D-DIA磷酸化蛋白质组学相比于常规的磷酸化蛋白质组学有什么优势?
A4:4D技术在原有保留时间、质荷比和离子强度的基础上,增加了离子淌度(CCS),能够显著提升蛋白修饰的鉴定能力,也因此4D-DIA磷酸化蛋白组能够提供对磷酸化蛋白更高深度的检测和更准确的定量。